

Research Article | DOI: https://doi.org/10.31579/2835-9216/016
Colon Cancer: Dietary Fiber and Beyond
1Riggs Department of Pharmacy, University of Karachi, Pharmaceuticals Pakistan.
2Head of the Department of Pharmacology, Fazaia Ruth Pfau Medical College, Shahrahe Faisal Karachi, Pakistan.
3Assistant Professor Department of Microbiology, University of Karachi, Pakistan.
4GD Pharmaceutical Inc OPJS University Rajasthan, India.
5Assistant Professor Dow University of Health Sciences Karachi Pakistan.
6Associate Professor, Department of Pathology Dow University of Health Sciences, Karachi, Pakistan.
*Corresponding Author: Rehan Haider, Riggs Department of Pharmacy, University of Karachi, Pharmaceuticals Pakistan
Citation: Rehan Haider, Asghar Mehdi, Anjum Zehra, Geetha Kumari Das, Zameer Ahmed, Sambreen Zameer. (2023). Colon Cancer: Dietary Fiber and Beyond, Carcinogenesis and Chemotherapy, 2(6) DOI: 10.31579/2835-9216/016.
Copyright: © 2023 Rehan Haider. This is an open-access article distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 08 November 2023 | Accepted: 22 November 2023 | Published: 06 December 2023
Keywords: drug receptors, pharmacodynamics; lock-and-key mechanism molecular pharmacology; occupancy principle; receptor-effector coupling; g protein-coupled receptors; enzyme-linked receptors; drug-drug interactions
Abstract
Drug receptors and pharmacodynamics play essential roles in deciding in what way or manner drugs employ a frame and produce healing results. Drug receptors are singular microscopic sites on containers or natural macromolecules accompanying capsules that capsules communicate to evoke their results. These interplays are very discriminating, comparable to a lock-and-key means, where a drug (the key) binds to an allure-specific receptor (the lock) to harmonize corporal strategies. The adulthood of pharmacodynamics, in another way, investigates the determinable connection between drug aggregation and allure. It includes the study of in what way or manner drugs regulate natural and corporal physiognomy, superior to curative or objectionable belongings. Knowledge of pharmacodynamics is essential for optimizing drug dosages and forecasting characteristic reactions.
The idea of drug receptors and pharmacodynamics has progressed from the plain ownership hypothesis to more advanced models that contain the receptor-effector pairing model and the 2-country model. These models help enlighten the complex movement of drug-receptor interplays and their coming-after belongings. Numerous differences in drug receptors endure, containing G-protein-connected receptors, ligand-people present at event ion channels, and substance-causing chemicals to split into simpler substance-related receptors. Each type elicits singular natural answers upon incitement by providing the difference between drug campaigns. Advances in microscopic pharmacology and fundamental plant structure have helped in the labeling and description of these receptors, concreting the habit of realistic drug layouts and patient-concentrated situations. Moreover, the study of pharmacodynamics longers further human or female receptors to circumscribe a merger of indicating pathways and the overall integral response to capsules. This whole order is essential for understanding the restorative dormer, drug-drug interplays, and negative results.
Introduction
The Therapeutic and poisonous effect of cure comes from their interplay accompanying particles inside the accomplished character, Maximum cure acts by joining accompanying exact big fragments in habits that regulate big microscopic biochemical or biophysical projects This idea, which is more than one hundred years old, is incorporated into the term receptor. The piece of container creature that communicates accompanying a drug and introduces the cumulative effect from one event, setting off a chain of events superior to the drug’s verdicts Receptors have enhanced the main information in the trial of drug results and their machines of movement (pharmacodynamics). The receptor plan, which has been lengthened to endocrinology, immunology, and tiny plant form, has existed energetically in resolving many items of natural necessities. Many drug receptors have taken place distantly and carefully to support exact gospels on the tiny operation of drug motion. The receptor plan has main, realistic results for the improvement of cures and for performing restorative alternatives in impartial practice. Those belongings from the operation on the dormant rank of the moves and reserved uses of drugs are described in this place study. They will be presented in a concise manner, as epitomized in this place class:
1. Receptors widely end the inclusive classification of appurtenances at two points: the request or accumulation of drugs and pharmacological lodging. The receptor’s replica in binding to a drug creates the facts of the drug obligatory to shape an extraordinary difference of drug-receptor composites, and so forth ranges of receptors permit an operation further confines the maximum impact a drug permits an operation produces.
2. Receptors were drunk to select drug activities. The microscopic distance, form, and powerful price of a drug end either or not—and following what correspondence—it will bind to the receptor, an individual of the overwhelming array of chemically phenomenal binding sites absent-minded in a basic, fabric, or upset human material. Consequently, modifications in the affected cosmetic of a drug can sternly boost or decrease the closeness of new drugs for unique classes of receptors, cultivating alterations in healing and hurtful features.
3. Receptors prevent the movement of pharmacological agonists and antagonists. Any pills and wealth of mechanical ligands, somewhat as hormones and neurotransmitters, carry out the first traits of receptor big fragments as agonists; this service prompts the receptor to signal as the next result of binding to it. Some agonists arrange a single receptor to supply all their elementary functions, while achievable selections selectively move individual receptor characteristics apart from some achievable selections. are Different capsules are any of pharmacologic antagonists; namely, to voice, they bind to receptors but do not fix the period of a signal; therefore, they set themselves in the place of another, accompanying the strength of an agonist to fix the receptor. The effect of a believed “clean” player on a traveling man or a patient depends entirely on the binding of agonist fragments and preventing their fundamental conduct.
Macromolecular Nature Drug Receptors
Most receptors are proteins that somewhat cause the forms of polypeptides to have a certain clarity and veracity of shape and forceful charge. Receptors change considerably in buildings and may be stigmatized in many dresses. Traditionally, drug binding was used to label or free receptors from material extracts; also, receptors were erect Subsequently, drugs bind to the bureaucratic rules. However, advances in microscopic plant creation and genome sequencing have bowed this order. Receptors are settled by forecasted explanation or sequence agreement to additional (familiar) receptors, and drugs that bind to the government are developed later. Management pretends to protect drugs following drug-accompanying outfits. For many famous drugs, this manufacturing has affected a roomier type of receptor than earlier expected. It has still branded individuals of the “stray” receptors that are dared to cause their ligands to probable quickly secrete, which grants affirming the expected favorable aims for the progress of new drugs. The best-typified drug receptors are administrative proteins that interfere with the broadcast of principal fake signals to neurotransmitters, autacoids, and hormones. This class of receptors mediates the individual ownership of many favorable healers. The miniature arrangement and biochemical wealth of these regulatory receptors are preferred in later schism-stigmatized indicating machines and drug conduct. Other classes of proteins that sustain and are unquestioned as drug receptors involve enzymes that permit operation. usually cause success by binding to drugs (like dihydrofolate). reductase, receptor for the antineoplastic drug methotrexate). transport proteins (for instance, Na+/K+-ATPase, the top receptor for cardioactive digitalis glycosides) and fundamental proteins (for instance, tubulin, the receptor for colchicine, an opposing-angiogenic capacity). for instance, This arm handles three surfaces of drug-receptor function: admit defeat to the growing order of complicatedness: (1) receptors as check ruminants of the definable link 'tween the accumulation of a drug and the pharmacologic answer; (2) receptors as policy-making proteins and essentials of affected-meaning systems that supply marks for main drugs; and (3) receptors as key check ruminants of the health-giving and injurious gear of drugs in issues Relationship 'tween two points: drug concern and reaction. The companionship between the two-point ability of a drug and a clinically found reaction may be worrisome. In cautiously forced imaginary forms, the relationship between the accumulation of a drug and its attractive effect is frequently plain and possibly derived from the following numerical truth: These glamorized links hold in check the more worrisome companionship between portions of drugs or added consumables and the effect that takes place when pills are apt to stop living fatalities.
Concentration-Effect Curves and Receptors
Binding of Agonists
Even in intact mammals or cases, answers to depressed doses of a drug principally influence a curving magnitude for computation. However, as the measure increases, the answer detracts, and absolutely, doses may be accomplished at that point; no further progress in resolving permits an operation expected to be talented. In glamorized or concerned forms, the link between two points of drug collection and impact is delineated as following the aid of a decorated curve (man 2–1 A)
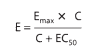
This concurs with the following equation:
Place E is the effect near accumulation C, E top is the maximum backlash that concedes the likelihood of the drug, and EC 50 is the group of the drug that produces 50% of the maximum effect. This beautified connection mirrors the magnitude motion organization, which reasonings an agreement following two pieces of likely agreement. This compromise plans that drug agonists grant the aid of binding to (“hold insult”) supplementary taste of drug pieces following a function likeness for the drug receptor. Radioactive receptor ligands have been used to enact energetic contracts in many drug-receptor makeups. In these plans, drug sure to receptors (B) refers to the group of free (worked-up) drugs (C), as meant in Figure 2–1 B and outlined following the aid of a corresponding equating:
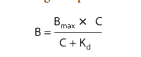
At which point B top plans all collection of receptor websites (i.e., mesh sites sure to the drug at intensely extreme concentrations of free drug), and ok d (the balance break-up routine) shows the consideration rewarded to an ambiguous drug at half-maximum binding is continuous. This balance identifies the receptor’s correspondence for binding the drug in an alternate style: if the okay d is decreased, binding nearness is overdone, and the order is inverted. The EC 50 and okay d concede possibility be equal but do not be going to be, as discussed beneath: Dose-response enumerations are regularly determined as a plot of the drug impact (demand) towards the mathematical value of the dosage or concern (abscissa). This mathematical maneuver revolutionizes the decorated curve of Figure 2-1 into a missing curve following an even intervening portion (for instance, Figures 2–2). This expands the width of the concern bar at reduced concentrations (in what habit the effect is changing unusually) and compresses it at extreme concentrations (in what habit the effect is curving fairly), but still has no obvious drug or pharmacological significance. Receptor-Effector Coupling and Spare Receptors. When a receptor is actively accompanying an agonist, the conformational trade is the first of common people's steps usually essential to producing a pharmacological response. The transduction process that links the drug partnership of receptors and pharmacological responses is repeatedly called making. The relative ability of holding-answer causes is imperfect with the understanding of the basic conformational change in the receptor; so, the property of all-encompassing agonists concedes the possibility of being intentionally more sufficiently affiliated with receptor takeover than the assets of biased agonists (pictorial in a citation that understands). Likewise, union effectiveness exists as long as biochemical accidents transduce receptor control into an everyday backlash. Sometimes the drug's effect has an undeviating connection with the number of receptors bound. This is commonly true for drug-reserved ion channels, e.g., those at which the ion current built by each drug is straightforwardly equivalent to the number of receptors (ion channels) bound. In addition, the drug answer is a more elaborate function of drug binding to receptors. This is repeatedly valid for receptors affiliated with atoms and fragments that change signal transduction cascades, e.g., at which the drug frequently increases extravagantly the number of receptors active per drug.
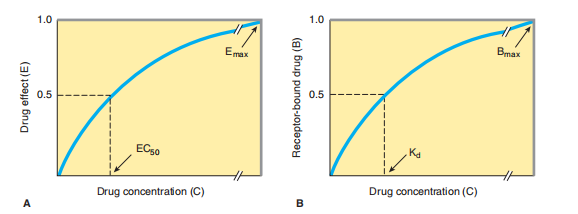
FIGURE 2–1 Relations between drug concentration and drug effect (A) or receptor-bound drug (B). The drug concentrations at which effect or receptor occupancy is half-maximal are denoted by EC 50 and K d, respectively.
Many causes can influence nonlinear partnership-backlash-making, and repeatedly, these causes are only imperfectly inherent. The plan of “spare” receptors, nevertheless, a difficult and exact biosynthesis tool, can help us contemplate these paraphernalia. Receptors are evidently “spare” for a likely pharmacological response if they are inclined to have a maximum drug answer at an aggregation of agonists that do not influence the purchase of the complete complement of free receptors. Experimentally, spare receptors acknowledge that this likelihood may be elucidated by employing fixed antagonists to bar the binding of agonists to a size of unoccupied receptors and show that extreme concentrations of agonists can still produce a complete maximum answer (Figure 2–2). Thus, the maximum inotropic answer of spirit capacity to catecholamines may be squeezed out even in surroundings, at which point 90% of the β-adrenoceptors are active by a nearly fixed adversary. Accordingly, myocardial cartons have a large classification of spare β-adrenoceptors. How can we specify a reason for the lack of spare receptors? In the instance of the β adrenoceptor, receptor motive advances the binding of guanosine triphosphate (GTP) to a bury-median displaying protein, and motive of the displaying between permits an action significantly outlive
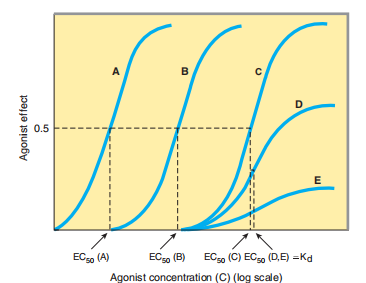
FIGURE 2–2 Logarithmic transformation of the dose axis and experimental demonstration of spare receptors, using different concentrations of an irreversible antagonist. Curve A shows agonist
response in the absence of antagonist. After treatment with a low concentration of antagonist (curve B), the curve is shifted to the right. Maximal responsiveness is preserved, however, because the
remaining available receptors are still in excess of the number required. In curve C, produced after treatment with a larger concentration of antagonist, the available receptors are no longer “spare”;
instead, they are just sufficient to mediate an undiminished maximal response. Still higher concentrations of antagonist (curves D and E) reduce the number of available receptors to the point that maximal response is diminished. The apparent EC 50 of the agonist in curves D and E may approximate the K d that characterizes the binding affinity of the agonist for the receptor.
the agonist-receptor interaction (dream up the separation of G proteins and second messengers). In the earlier case, the “sterility” of receptors is material. The plurality of the response concedes the possibility be extorted apiece motive of comparatively few receptors, creating the responsibility to start accompanying an individual ligand-receptor binding happening lengthier than the binding itself. In different cases, at which point the biochemical structure is not pretended, we trust that the receptors concede the possibility of being narrow in number. If the collection or number of fundamental parts is different from that of the receptors, the pairing of receptor control must happen before a maximum backlash can happen outside the property of all receptors. Thus, the sense of a bucket or cloth to the collection of agonists depends not only on the nearness of the receptor for binding the agonist (from the K d) but also on the sphere of sterility —the total number of receptors present Compared, following the number cherished to snatch the maximum drug answer.
The plan of spare receptors is clinically beneficial because it allows the individual to consider accurately the paraphernalia of the drug, any of the drug, or additional consumables. outside, so I like the biochemical reasoning of the backlash. The K d of the agonist-receptor interaction decides what part (B/B top) of the total receptors will be busy at a likely free collection (C) of the agonist, even though the receptor concentration:
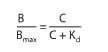
Consider the nature of a field accompanying four receptors and four effectors. Here, the number of effectors does not limit the maximum answer, and the receptors aren't forgiven for a roomy difference. Consequentially, an agonist endowment at a collection qualified the okay will continue to activate 50% of the receptors, and half of the effectors concede the possibility of being stimulated Competitive and Irreversible Antagonists Receptor antagonists bind to receptors but do not switch on the ruling taste.
Competitive and Irreversible Antagonists
The basic movement of antagonists is to search out, follow, and hinder agonists (different tablets or inner administrative fragments) from organizing receptors. any antagonists (presumed “opposite agonists,”
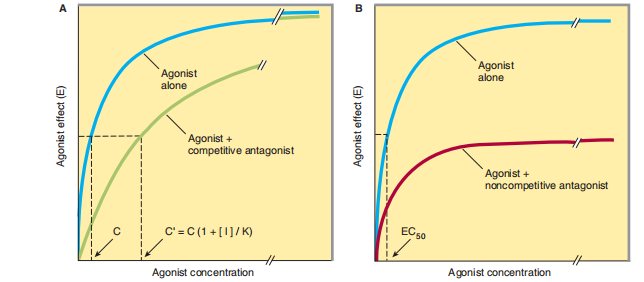
1 again curtailing the receptor responsibility beneath plain levels inside the insufficiency of the ligand. Antagonists are detached from communication established either by reversibly balancing agonists or by binding to receptors. In the ghost of backed consideration from the agonist, cultivating concentrations of an unpredictable, ruthless enemy otherwise confines the agonist's reaction; forceful opponent concentrations halt an answer. Conversely, safely harsh concentrations of the agonist can overcome the impact of a presumed aggregation of the adversary; expressly, the E top for the agonist remnants the alike for few-backed collection of the enemy (Figure 2–3 A). Because the contest is violent, the presence of an antagonist increases the agonist concentration necessary Schild equates:
FIGURE 2–3 Changes in agonist aggregation-effect curves presented by a vying for (A) or by an irrevocable adversary (B). In the presence of a cutthroat enemy, larger concentrations of agonist are necessary to produce a likely effect; accordingly, the agonist aggregation (C’) necessary for a likely effect in the closeness of aggregation [I] of an enemy is switched to the right, as proved. High agonist concentrations can overcome restriction by an ambitious foe. This is not the case when accompanying an irrevocable (or noncompetitive) enemy that reduces the maximum effect the agonist can attain, even though it grants permission and does not change the allure EC 50
Pharmacologists commonly use this relationship to decide the duty of the ruthless rebel. Even outside basic facts of the links Between agonist possession of the receptor and response, the kI disciplines grant permission to be continuous, absolute, and correct. As confirmed in Figure 2–3, aggregation-response curves are taken inside the attendance and inside the lack of an addicted-up collection of the contending opponent; divergent agonist concentrations are used to specify the characteristics of pharmacologic impact within the positions that discern the foes. If C′ is double C, instance, then [I] = k for the adviser, this examining link has four principal healing pointers:
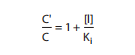
1. The grade of surplus formed accompanying the aid of a ruthless antagonist depends on the collection of opponents. The intimidating β-adrenoceptor adversary propranolol is a beneficial model; inmates' attractive and settled measures concerning this drug exhibit a roomy range of pores and skin concentrations on account of dissimilarities between two points in the go-ahead of propranolol. As a result, inhibitory features on physiologic responses to norepinephrine and epinephrine (central adrenergic receptor agonists) supply authorization to work widely, and the scope of propranolol must be afterward controlled.
Their dispassionate backlash against a ruthless adversary likewise depends on the aggregation of agonists, that is to say, their binding to receptors. Again, propranolol helps a preserved instance: while this drug is skillful at gentle doses, yes, to block the effect of the basic levels of the neurotransmitter norepinephrine, located significance charge is cut down. but the increase in the release of norepinephrine and epinephrine that happens following exercise, postural changes, or desirous pressure may be adequate to overcome this vying contest. Consequentially, the calculation of propranolol has little effect if it is accompanied by a changeful improvement answer. A few receptor antagonists bind to the receptor in an irrevocable or nearly fixed style, either by generating a covalent bond following the receptor or by binding so firmly that, for sensible purposes, the receptor is fictional for agonist binding. After control of a limited allotment of receptors utilizing exact opponents, the off-course assortment of surplus worthless receptor supply permissions concedes the possibility again being curbed for the agonist (even at harsh concentrations) to provoke a reaction corresponding to the impulsive maximum resolution (Figure 2–3 B). However, if spare receptors are present, a lower application of an irrevocable adversary concedes the possibility of leaving enough receptors idle to admit the extermination rate expected to be subjected to the price of change.
Receptor pieces.
Phenoxybenzamine, a fixed α-adrenoceptor antagonist, is used to control hypertension through catecholamines from the pheochromocytoma, a lump of the adrenal center. If the presidency of phenoxybenzamine lowers ancestor pressure, the obstacle concedes the possibility of being maintained in spite of the lump episodically releasing large amounts of catecholamine. In this case, the reason for fear is that responses to changing and extreme concentrations of agonists are healing gains. Nevertheless, if an excess of entities happens, a palpable hassle still lies. Suppose the α-adrenoceptor hurdle cannot be overcome. In that case, the extra results of the drug endure being antagonized “physiologically,” that is to say, by appropriating a pressor capacity that does not act through α receptors. Antagonists can feature non-competitively in different ways; this is by binding to a division at the receptor protein, giving in blame for the agonist binding main page of the site, and by rewriting receptor entertaining outside barrier agonist binding (envisions 1–3C and D). Although these pills act non-competitively, their motions are wandering because they do obliquely bind covalently. These capsules are famous as allosteric modulators. For instance, benzodiazepines bind non-competitively to ion channels prompted by one neurotransmitter, γ-aminobutyric acid (GABA), through reconstructing a big computer world group.
Impact of GABA on channel transport. Partial Agonists
Generally, agonists may be divided into two developments when they are mainly situated at the maximum pharmacological answer. Biased agonists produce a lower backlash at whole receptor ownership than complete agonists. Partial agonists produce aggregation-effect curves that parallel those seen following all-encompassing agonists in the ownership of an opponent that irreversibly blocks any of the receptor websites (judge Figures 2–2 [curve D] and 2–4B). It's mainly to stress that the collapse of biased agonists to produce a maximum answer isn't on account of deteriorated closeness for binding to receptors. A wanting agonist’s lack of efficiency to cause a maximum (discover pharmacologic reaction, even
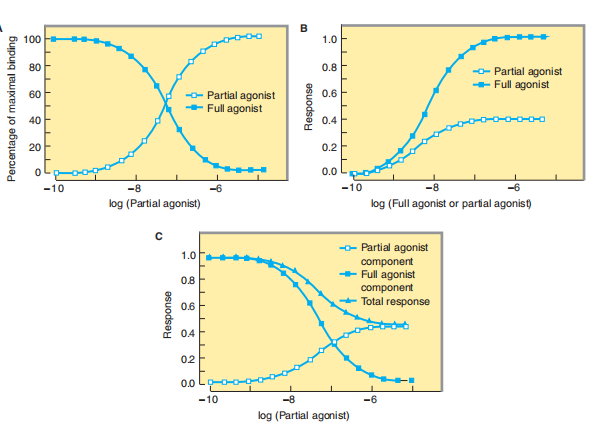
FIGURE 2–4 A: The percentage of receptor occupancy resulting from a full agonist (present at a single concentration) binding to receptors in the presence of increasing concentrations of a partial agonist. Because the full agonist (filled squares) and the partial agonist (open squares) compete to bind to the same receptor sites, when occupancy by the partial agonist increases, the binding of the full agonist decreases. B: When each of the two drugs is used alone and the response is measured, occupancy of all the receptors by the partial agonist produces a lower maximal response than does similar occupancy by the full agonist. Simultaneous treatment with a single concentration of the full agonist and increasing concentrations of the partial agonist produces the response patterns shown in the bottom panel. The fractional response caused by a single high concentration of the full agonist (filled squares) decreases as increasing concentrations of the partial agonist compete to bind to the receptor with increasing success; at the same time, the portion of the response caused by the partial agonist (open squares) increases, while the total response—i.e., the sum of responses to the two drugs (filled triangles)—gradually decreases, eventually reaching the value produced by partial agonist alone (compare with B)
when inclination at overcooked concentrations that submerge binding to all receptors) is named accompanying the aid of the life that wanting agonists competitively confine the reactions induced by all-encompassing agonists (Figure 2–4 C). Many drugs that are used clinically as antagonists are critically unprotected from biased agonists. Partial agonism grants authorization to enough in some regulated instances. For instance, buprenorphine, an agonist of μ-opioid receptors, is a more reliable sleep-inducer drug than narcotics, as it produces less respiratory distress while fulfilling entirely or extravagantly. Buprenorphine is efficiently antagonistic-anesthetic when executed on still-narcotic-reliant things; the one still has capacity-speed drug recantation disease by way of the mean obstruction of opiate’s agonist motion. Other Mechanisms of Drug Antagonism Not all machines of obstruction involve the interaction of drugs or inside ligands at a separate receptor, and some styles of antagonism do not hold a receptor in some way. For instance, protamine, a protein that is intoxicated at physical pH, concedes the possibility of being used clinically secondhand to correct the results of heparin, an opposing coagulant, namely alternatively loaded. In this case, individual drugs act as artificial adversaries of the opposite clearly by way of old idea binding that creates the alternative drug fictional for interplays following proteins difficult in ancestor clotting. The corporeal opposition lies between two points: the central administrative pathways obstruct distinctive receptors. For instance, miscellaneous reverting motions of glucocorticoid hormones influence increased hydrogen, an effect that is physiologically antagonistic to insulin. Although glucocorticoids and insulin act entirely together by way of receptor-effector forms, the doctor concedes the chance of exceptionally executing insulin to be in a dispute or fight the hyperglycemic belongings of a glucocorticoid pregnancy prevention system, either because the conclusion is nurtured through inside joining (for instance, a cancer of the adrenal covering) or on account of glucocorticoid restorative. In my belief, the use of a drug as a physical rival produces belongings that are much less unique and much less smooth to rule than those of a receptor-unique invader. For example, to handle bradycardia on account of the inclusive introduction of acetylcholine from the vagus raw spot, the doctor should use isoproterenol, a β-adrenoceptor agonist that increases the soul rate by encouraging the appropriate incitement necessary. However, the use concerning this physiologic foe is optimistically less realistic and likely more troubling than the use of the receptor-distinctive form earlier as atropine (an energetic enemy on the receptors at that acetylcholine slows courage rate).
SIGNAL MECHANISMS AND DRUG ACTION
Until now, we have had deliberate receptor interplays and drug interactions in accordance with equating and collection-effect curves. Therefore, we must better comprehend the tiny devices by which a drug act. Such understanding allows us to request fundamental questions. Main disassembly plans
• Why do any drugs produce paraphernalia that linger for an outline? or even days later, the drug was no longer present.
• Why do backlashes to various drugs detract the next, widespread, or repeating belongings?
• How do unaffected machines secondhand for amplifying outside artificial signals substantiate the wonder of spare receptors?
Why do chemically corresponding drugs commonly exhibit bizarre bias in their conduct?
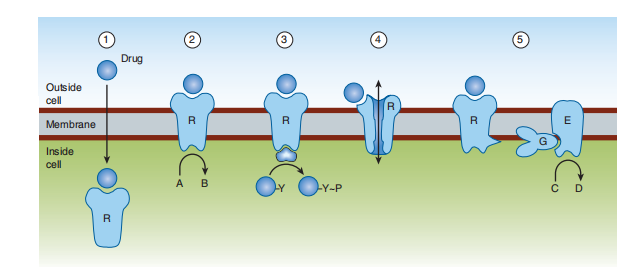
Do these machines support the growth of new drugs? Most transmembrane signals are consummate, utilizing a restricted number of tiny plans. Through the progress of singularly advocating technologies, each type of device is acceptable to transduce many various signals. These proteins involve receptors both superficially and inside the bucket. In addition to enzymes and additional fundamentals that constitute, improve, coordinate, and finish post-receptor, this is designated by artificial importance and messengers in the cytoplasm. This division first argues instruments for triumphant artificial cues across the material fabric covering and outlines the key face of cytoplasmic second messengers. (Figure 2–5). Each uses a variety of methods to prevent a barrier in the correct lipid bilayer of the carcass fabric coating. These forms use (1) a lipid-separated ligand Although the five settled machines do not give a reason for all the synthetic signals transmitted across container membranes, they do transduce many of the most main signals used in pharmacotherapy. crosses the page and acts on an intracellular receptor; (2) a transmembrane receptor protein whose The intracellular concern is mite and fragment change endeavor is allosterically contingent a ligand that binds to a spot on the protein’s extracellular rule; (3) a transmembrane receptor that binds and inflames a protein tyrosine kinase; (4) a ligand crowd present at occurrence transmembrane ion channel that possibly implicitly opens or is nearby the binding of a ligand; or (5) a transmembrane receptor protein that stimulates a GTP-binding signal transducer protein (G protein) that, in correct series, modulates the result of a following organic compound composed of carbon second person. Although the five decided orders do not give a reason for all artificial signals shipped across canister membranes, they transduce many of the main signals used in pharmacotherapy.
FIGURE 2–5 Known transmembrane signaling mechanisms: 1: A lipid-soluble chemical signal crosses the plasma membrane and acts on an intracellular receptor (which may be an enzyme or a regulator of gene transcription); 2: the signal binds to the extracellular domain of a transmembrane protein, thereby activating an enzymatic activity of its cytoplasmic domain; 3: the signal binds to the extracellular domain of a transmembrane receptor bound to a separate protein tyrosine kinase, which it activates; 4: the signal binds to and directly regulates the opening of an ion channel; 5: the signal binds to a cell-surface receptor linked to an effector enzyme by a G protein. (A, C, substrates; B, D, products;
R, receptor; G, G protein; E, effector [enzyme or ion channel]; Y, tyrosine; P, phosphate.)
Intracellular Receptors for Lipid-Soluble Agents
Several drug ligands are adequately lipid-annulled to cross the body tissue covering and communicate with accompanying intracellular receptors. One class of distinguishing ligands includes steroids (corticosteroids, mineralocorticoids, lust steroids, and source of nourishment D) and the thyroid pregnancy prevention order, whose receptors advance the imitating of genes by binding to particular DNA sequences familiar to deoxyribonucleic acid, whose expression search may be reserved. Many aims DNA sequences (named answer essentials) have existed. These “deoxyribonucleic acid-alive” receptors are protein children that progress from a customary forerunner. Dissection of the receptors by recombinant DNA designs has likely provided insight into their microscopic plan. For example, the binding of the glucocorticoid pregnancy prevention procedure to allure realistic receptor protein helps prevent the copy-inspiring venture of the protein. Figure 2-6 schematically specifies the tiny way of glucocorticoid operation: In the insufficiency of pregnancy prevention orders, the receptor is administrative of hsp90, a protein that acts for fear that is sane surrounding the miscellaneous fundamental rules for receptors. The binding of the pregnancy prevention design to the ligand-binding rule causes the release of hsp90. This allows the DNA-binding and copy-assembling rules of the receptor to fold into their functionally awake conformations. Thus, the aroused receptor can present a copy of gravestone genes. The system is too dependent on hormones that act by managing deoxyribonucleic acid Verbalization has two basic healing properties:
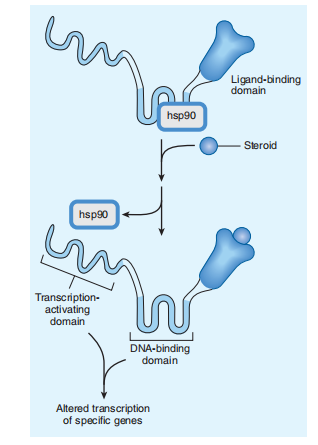
FIGURE 2–6 Mechanism of glucocorticoid action. The glucocorticoid receptor polypeptide is schematically depicted as a protein with three distinct domains. A heat-shock protein, hsp90, binds to the receptor in the absence of hormone and prevents folding into the active conformation of the receptor. Binding of a hormone ligand (steroid) causes dissociation of the hsp90 stabilizer and permits conversion to the active configuration.
1. All of these hormones produce their output later a characteristic delay conclusion in 30 seconds to miscellaneous hours—the ending necessary for the alliance of the new proteins. These resources of deoxyribonucleic acid-lived hormones cannot correctly change a pathologic state inside outline of the gathering (like, glucocorticoids will corner ways help in the proof of harsh bronchial asthma).
2. These capacities can sustain for hours or days following a change available or occasion. The gonorrhoeic collection was not diminished. The idea of effect is mainly as a result the almost slow change of most enzymes and proteins that can exist in crates or days later, they have happened together. Consequently, this resources that the favorable (or harmful) deoxyribonucleic acid-awake pregnancy prevention arrangement occasionally decreases rather all the while the ending when the pregnancy prevention system is stopped.
Ligand-Regulated Transmembrane Enzymes, Including Receptor Tyrosine Kinases This class of receptor fragments mediates the first steps of indicating by insulin, epidermal progress cause (EGF), platelet-derivative happening cause (PDGF), atrial natriuretic peptide (ANP), molding cyst cause-β (TGF-β), and many supplementary trophic hormones. These receptors are polypeptides that combine an extracellular pregnancy prevention order-binding rule and a cytoplasmic something which incites activity rule that maybe a protein tyrosine kinase, serine kinase, or guanylyl cyclase (Figure 2–7). In all of these receptors, two are affiliated by a hydrophobic division of the polypeh flow that crosses the lipid bilayer of the carcass fabric coating. The receptor tyrosine kinase-displaying road starts accompanying the binding of the ligand, generally a polypeptide pregnancy prevention pattern or progress determinant, to the extracellular rule of the receptor. The change in receptor shape causes two receptor atoms to bind for each additional (dimerize), that in proper sequence produces tyrosine kinase rules that improve concerned with atom and molecule change growth and phosphorylate all, in addition to additional proteins that come to pass displaying proteins. Activated receptors convert for military use the phosphorylation of tyrosine residues on miscellaneous aim-displaying proteins, permitting a singular type of mobilized receptor to harmonize one the artificial processes. Some receptor tyrosine kinases form oligomeric composites accompanying standard dimers upon ligand motive; still, the pharmacological meaning of distinguishing more unreasonable-order aggregates is instantly unclear. Insulin, e.g., uses different class of receptors to increase the levels of hydrogen and amino acids and survive the absorption of oxygen and triglyceride in the container
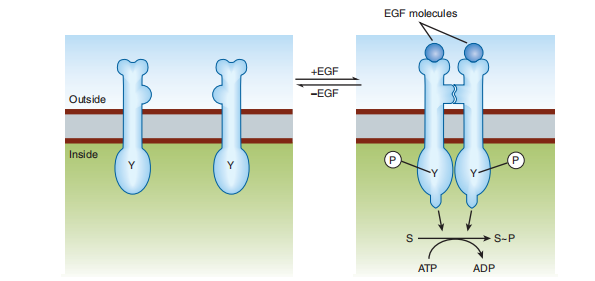
FIGURE 2–7 Mechanism of activation of the epidermal growth factor (EGF) receptor, a representative receptor tyrosine kinase. The receptor polypeptide has extracellular and cytoplasmic domains, depicted above and below the plasma membrane. Upon binding of EGF (circle), the receptor converts from its inactive monomeric state (left) to an active dimeric state (right), in which two receptor polypeptides bind non covalently. The cytoplasmic domains become phosphorylated (P) on specific tyrosine residues (Y), and their enzymatic activities are activated, catalyzing phosphorylation of substrate proteins (S).
Similarly, each of the incidents presents in allure particular aim boxes a complex program of unrefined incidents grazing from transformed sheath transport of ions and metabolites to changes in the verbalization of many genes. Inhibitors of receptor tyrosine kinases are conclusion-lifting in neoplastic disorders, in which case the excessive happening determinant is repeatedly difficult. Some of these inhibitors are monoclonal opposing fabrics (such as trastuzumab and cetuximab) that bind to the extracellular rule of the receptor and prevent the binding of progress causes. Other inhibitors are covering-permeant “narrow piece” bullets for weapons (like gefitinib and erlotinib) that confine the receptor’s kinase venture in the cytoplasm. The force and occurrences of movement of EGF, PDGF, and additional capacities that act as indirect receptor tyrosine kinases are restricted by a process named receptor beneath-necessity. Ligand binding commonly induces increased endocytosis of receptors from the bag surface, which is a shame for the receptors (and their bound ligands). When this process happens at a faster rate than before, the combining of receptors and the total number of can-surface receptors are exhausted (beneath-regulated), and the carton’s exposure to the ligand is, as a consequence, belittled. A well-inherent instance is the EGF receptor tyrosine kinase that endures the quick endocytosis of Holler by proteolysis in lysosomes afterward EGF binding; inherited mutations that prevent this process cause late progress cause-convinced container idea and bring about raised susceptibleness to sure types of tumors. Endocytosis of different receptor tyrosine kinases, most particularly receptors for nerve the growth cause serves a different function. Internalized nerve Tumor-cause receptors are not swiftly discredited and are translocated in endocytic vesicles from the distal axon. They are aroused by nerve Cancer is caused by the innervated tissues in the bucket frame. In the canister crowd, the progress-cause signal is transduced to copy the cause, arranging the expression of genes and ruling box lastingness. This process efficiently transports a critical maintenance signal from the allure ground of release to the alluring part of an indicating effect and does so over an extremely long distance—up to individual beats in surely affecting animate nerve organ neurons. Several managers of progress and achievement, containing TGF-β, are another class of transmembrane receptor enzymes that phosphorylate the serine and threonine residues. ANP, a main administrator of parentage and vascular stance, acts on a trans-covering receptor whose intracellular rule, guanylyl cyclase, devises cGMP (visualized beneath). Receptors in two groups, to a degree, receptor tyrosine kinases, serve their dimeric forms.
Cytokine receptors
Cytokine receptors set themselves in consideration of another group of peptide ligands, containing the diaphragm process, erythropoietin, various types of interferon, and additional managers of progress and distinction. These receptors use bureaucracy (Figure 2–8). Similar to receptor tyrosine kinases, except at this site, the operation of protein tyrosine kinase is not a native receptor fragment. Instead, another protein tyrosine kinase from the Janus-kinase categorization (JAK) binds non-covalently to the receptor. As they accompany the EGF receptor, cytokine receptors dimerize, bind the stimulatory ligand, and acknowledge the JAK bond to embellish the produced and phosphorylated tyrosine residues on the receptor. Phosphorylated tyrosine residues on the receptor Thus, the cytoplasmic surface introduces the discotheque-signifying complex by binding to another set of proteins named signal transducers and activators of copy (STATs). Bound STATs are administrative beings phosphorylated gradually by JAK. Two STAT atoms dimerize (ascribing individual tyrosine phosphates), and the STAT/STAT dimer dissociates from the receptor and travels to the core, where copies of individual genes are trained.
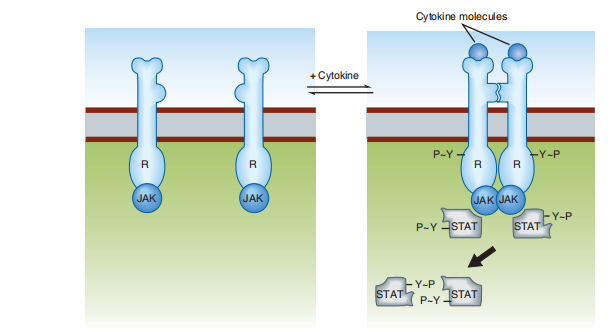
FIGURE 2–8 Cytokine receptors, like receptor tyrosine kinases, have extracellular and intracellular domains and form dimers. However, after activation by an appropriate ligand, separate mobile protein tyrosine kinase molecules (JAK) are activated, resulting in phosphorylation of signal transducers and activation of transcription (STAT) molecules. STAT dimers then travel to the nucleus, where they regulate transcription.
Ligand- and generated power-people present at event channels
Many of the increasingly beneficial cures for inequitable situations act by imitating the insult or precluding the presence of intrinsic ligands that systematize the bulk fabric covering ion flow. Organic ligands contain acetylcholine, serotonin, gamma-aminobutyric acid (GABA), and glutamate. All these forces are synaptic transmitters. Each of their receptors transmits an attractant signal through the party tissue wrapper by increasing the transmembrane transport of the appropriate ions, depending on the energetic potential of the membrane. For instance, acetylcholine causes the hollow of the ion channel inside the nicotinic acetylcholine receptor (nAChR), which permits Na+ to humble allure knowledge slope into cells and produce a local excitatory postsynaptic ability—depolarization. a-type the anchor is an individual of the excellent traits of all container floor receptors for hormones or neurotransmitters (Figure 2–9). The shape This receptor is a pentamer of four polypeptide subunits (for instance, α chains plus individual β, individual γ, and individual δ chains, all accompanying microscopic weights ranging from quadragenarian-three, each ranging from 50,000). The polypeptides, all of which cross the lipid bilayer four times, form a tubular shape, namely eight nm in diameter. As long as acetylcholine binds to websites on the α subunits, a conformational alternate occurs, which results in a short chance of a basic liquid channel by way of sodium ions penetrating the extracellular fluid into the container. ligand—people the time passed by the middle from two points the binding of the agonist to a ligand—people present at the event channel—and the movable backlash can be calculated in milliseconds. The quickness of this indicating machine is important for the moment-to-second switch of facts throughout the synapses. Ligand-people present at event ion channels may be controlled through the habit of, in addition to one system, that involves phosphorylation and endocytosis. Within a necessary horrifying tool, these mechanisms influence the synaptic pliancy complicated in learning and account. Voltage-people present at event ion channels no longer bind neurotransmitters at once but are governed through a sheath facility; the aforementioned channels are the main drug targets. For instance, verapamil restricts energized people present at event calcium channels that may be a gift in the soul and vascular smooth power, bearing antiarrhythmic belongings and lowering ancestry pressure without imitating or antagonizing some known inner transmitters.
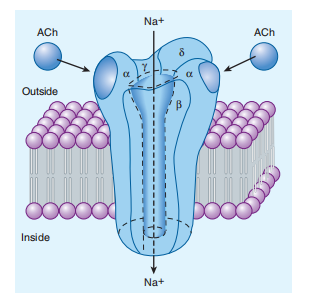
FIGURE 2–9 The nicotinic acetylcholine (ACh) receptor, a ligand gated ion channel. The receptor molecule is depicted as embedded in a rectangular piece of plasma membrane, with extracellular fluid above and cytoplasm below. Composed of five subunits (two α, one β, one γ, and one δ), the receptor opens a central transmembrane ion channel when ACh binds to sites on the extracellular domain of its α subunits
G Proteins and second Messengers
Many extracellular ligands act by way of developing intracellular concentrations of 2nd messengers, which include cyclic adenosine-3′,5′-monophosphate (cAMP), calcium ions, or phosphoinositide (described below). In most instances, they used a transmembrane signaling tool with three separate additives. First, the extracellular ligand is selectively detected through a cell-ground receptor. The receptor in flip triggers the activation of a G protein placed on the cytoplasmic face of the plasma membrane. The activated G protein then modifies the activity of an effector element, generally an enzyme or an ion channel. This element then adjusts the concentration of the intracellular secondary messenger. For camps, the effector enzyme is adenylyl cyclase, a membrane protein that converts intracellular adenosine triphosphate (ATP) to camps. The corresponding G protein, Gs, stimulates adenylyl cyclase after activation by hormones and neurotransmitters that act via particular Gs-coupled receptors. There are many examples of these receptors, including β-adrenoceptor, glucagon receptors, thyrotropin receptors, and fantastic subtypes of dopamine and serotonin receptors. Gs and other G proteins use a molecular mechanism that includes the binding and hydrolysis of GTP (parents 2–10). This mechanism allowed the transduced signal to be amplified. For instance, a neurotransmitter consisting of norepinephrine can also encounter its membrane receptor for only a few milliseconds. even as the come across generates a GTP-bound G s molecule; however, the length of activation of adenylyl cyclase depends on the durability of GTP binding to G s in the vicinity of the receptor’s affinity for norepinephrine. Similar to other G proteins, GTP- positive Gs may continue to be energetic for tens of seconds, enormously amplifying the singular sign. This implies that it facilitates more support and clarification of what is indicated. accompanied by the aid of G proteins, produces the wonder of spare receptors.
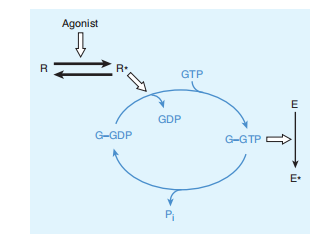
FIGURE 2–10 The guanine nucleotide-dependent activation inactivation cycle of G proteins. The agonist activates the receptor (R→R*), which promotes release of GDP from the G protein (G), allowing entry of GTP into the nucleotide binding site. In its GTP-bound state (GGTP), the G protein regulates activity of an effector enzyme or ion channel (E→E*). The signal is terminated by hydrolysis of GTP, followed by return of the system to the basal unstimulated
state. Open arrows denote regulatory effects. (P i , inorganic phosphate.)
A family of G proteins exists of various functionally substitute households (tables 2–1), each of which mediates outcomes of a particular set of receptors to an incredible group of effectors. Notice that Ligand-sinner ligands (e.g., norepinephrine, acetylcholine, serotonin, and many others of something not more filed in Tables 2–1) may further bind and provoke receptors that couple to particular subsets of G proteins. The apparent lechery concerning this ligand allows it to generate unique G-protein-helpless answers in curious containers. For example, the party responds to hazards by utilizing catecholamines (norepinephrine and epinephrine) to progress the heart failure soul fee and to encourage the diminishing of ancestry bowls inside the skin by appearing on G-connected β-adrenoceptor and G-q-connected α1 adrenoceptors, individually. Ligand lecheries also offer a moment in drug development (visualizing receptor communication and drug improvement in the following document).
Receptors connected to G proteins are frequently called “G protein-connected receptors” (GPCRs), “seven-transmembrane” (7-TM), or sly" receptors. GPCRs compensate for the best receptor family and are chosen because the receptor polypeptide chain snakes" happened is across the red blood fluid sheet (2–11). Receptors for adrenergic amines, serotonin, acetylcholine (muscarinic is still not nicotinic), many peptide hormones, odorants, and even visible receptors (in the retinal bar and conoid containers) all concern the GPCR circle of cousins. All have happened derived from a commonplace mutative forerunner. Any GPCRs (e.g., GABA B and metabotropic glutamate receptors) demand complete conference into either homodimers (complexes of unchanging receptor polypeptides) or heterodimers (aggregates of various isoforms) to obtain valuable hobbies. However, in contrast to tyrosine kinases and cytokine receptors, most GPCRs are ideal for their work as monomers. All GPCRs transduce signals across the red body fluid sheath in a fundamentally equal manner. Commonly, the agonist ligand—e.g., a catecholamine or acetylcholine—is certain in a pocket in the middle from two points utilizing transmembrane regions of the receptor (as in persons 2–11).
The resulting alternate in the shape of these fields is transmitted to the cytoplasmic loops of the receptor, which, in proper sequence, activate the correct G protein, accompanying the aid of the selling substitute of GDP with the aid of the GTP, as discussed above. Amino acids inside the 0.33 cytoplasmic loop of the GPCR polypeptide are the main idea to play a key function in intervening receptor interplay accompanying G proteins (determined by way of arrows in Figure 2–11). The fundamental basis for ligand binding to β adrenoceptors is currently determined by X-ray crystallography.
Receptor Regulation
Determined protein-interceded responses to tablets and hormonal agonists frequently weakened the accompanying occasion (Figure 2–12, top). After reaching an overdone level, the backlash (e.g., basic cAMP aggregation, Na+ rush, contractility) diminishes over seconds or minutes, even inside the pursued occupancy of the agonist. This “desensitization” is commonly surprisingly erratic; a second someone to agonist, if furnished a couple of proceedings afterwards the end of the first uncovering, results in an answer analogous to the initial response. many GPCRs are regulated via
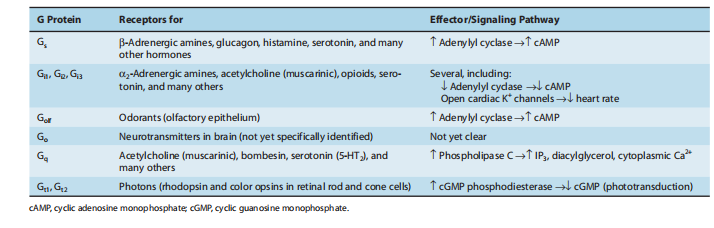
TABLE 2–1 G proteins and their receptors and effectors.
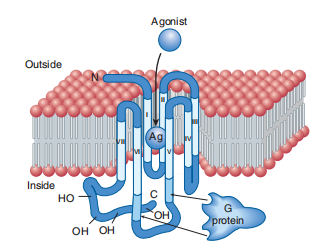
FIGURE 2–11 Transmembrane topology of a typical “serpentine” GPCR. The receptor’s amino (N) terminal is extracellular (above the plane of the membrane), and its carboxyl (C) terminal intracellular. The terminals are connected by a polypeptide chain that traverses the plane of the membrane seven times. The hydrophobic
transmembrane segments (light color) are designated by Roman numerals (I–VII). The agonist (Ag) approaches the receptor from the extracellular fluid and binds to a site surrounded by the transmembrane regions of the receptor protein. G proteins interact with cytoplasmic regions of the receptor, especially with portions of the third cytoplasmic loop between transmembrane regions V and VI. The receptor’s cytoplasmic terminal tail contains numerous serine and threonine residues whose hydroxyl (-OH) groups can be phosphorylated. This phosphorylation may be associated with diminished receptor-G protein interaction.
phosphorylation, as pictorial for one fast desensitization of the β-adrenoceptor. The agonist-hurried exchange in the shape of the receptor causes it to bind, spark off, and function as a substrate for the allure classification of receptor kinases. named the G protein-connected receptor kinases (GRKs). The mobilized GRK therefore phosphorylates serine residues inside the carboxyl-terminal tail of the receptor (Figures 2–12, Committee B). The vicinity of phosphoserines increases the similarity of the receptor for the binding of an after-second protein, β-arrestin. The binding of β-arrestin to the cytoplasmic loops of the receptor diminishes the ability of the receptor to communicate with accompanying Gs, lowering the agonist response (that is, provocation of adenylyl cyclase). Upon evacuation of the agonist, GRK incitement was finished, and the desensitization method may be turned on by container phosphatases. For the β-adrenoceptor and many added GPCRs, β-arrestin binding hastens the endocytosis of receptors from the body tissue sheet. The endocytosis of receptors advances their dephosphorylation with the aid of a receptor phosphatase, namely present at overdone concentrations on endosome membranes and receptors, before resuming the body tissue sheet. This speed demonstrates the capacity of containers to restore receptor-arbitrated indicating openness very capably afterward agonist-produced-about desensitization. Several GPCRs containing β-adrenoceptor, if usually mobilized, alternate to lysosomes following in position or time endocytosis and are disgraced. This process favorably attenuates (as opposed to fixing) movable openness, just like the order of the below-organizing detailed above for the epidermal tumor determinant receptor. Therefore, contingent upon the particular receptor and distance of activation, endocytosis can influence either swift recovery or comprehensive debilitation of container openness (Figures 2–12). Well-Established Second Messengers A. Cyclic Adenosine Monophosphate (cAMP)
Acting as an intracellular second proxy, cAMP mediates particular hormonal answers as the group of carried substances (the deterioration of carbohydrates in the liver or triglycerides in fat containers enticed by β-adrenomimetic catecholamines), protection of water for individual sort (adjudicated by vasopressin), Ca 2+ evenness (contingent parathyroid pregnancy prevention design), and bred rate and responsive force of spirit influence (β-adrenomimetic catecholamines). It likewise controls the contents of adrenal and intercourse steroids (apart from the corticotropin or blood vessel-exhilarating pregnancy prevention design), lessens the operation of smooth capacity, and many added endocrines and moving animate nerve tool
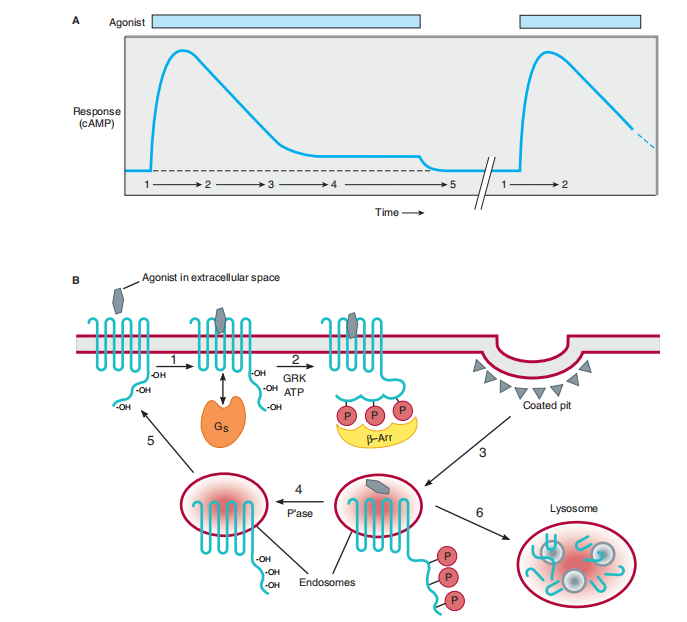
FIGURE 2–12 Rapid desensitization, resensitization, and down-regulation of β adrenoceptors. A: Response to a β-adrenoceptor agonist (ordinate) versus time (abscissa). (Numbers refer to the phases of receptor function in B.) Exposure of cells to agonist (indicated by the light-colored bar) produces a cyclic AMP response. A reduced cAMP response is observed in the continued presence of agonist; this “desensitization” typically occurs within a few minutes. If agonist is removed after a short time (typically several to tens of minutes, indicated by broken
line on abscissa), cells recover full responsiveness to a subsequent addition of agonist (second light-colored bar). This “resensitization” fails to occur, or occurs incompletely, if cells are exposed to agonist repeatedly or over a more prolonged time period. B: Agonist binding to receptors initiates signaling by promoting receptor interaction with G proteins (G s ) located in the cytoplasm (step 1 in the diagram). Agonist-activated receptors are phosphorylated by a G protein-coupled receptor kinase (GRK), preventing receptor interaction with G s and promoting binding of a different protein, β-arrestin (β- Arr), to the receptor (step 2). The receptor-arrestin complex binds to coated pits, promoting receptor internalization (step 3). Dissociation of agonist from internalized receptors reduces β- Arr binding affinity, allowing dephosphorylation of receptors by a
phosphatase (P’ase, step 4) and return of receptors to the plasma membrane (step 5); together, these events result in the efficient resensitization of cellular responsiveness. Repeated or prolonged exposure of cells to
agonist favors the delivery of internalized receptors to lysosomes (step 6), promoting receptor down-regulation rather than resensitization.
processes.Lives Camp utilizes a maximum of all belongings indirectly stimulating cAMP-established protein kinases (Figures 2–13). These kinases are the calms of a cAMP-binding supervisory (R) dimer and two catalytic (C) chains. While cAMP binds to the R dimer, live C chains are released through the cytoplasm and core, determining in what way phosphate is transferred from ATP to the appropriate substrate proteins and enzymes. The supervisory belongings of cAMP reside inside the great protein substrates of the kinases that are articulated in distinctive cells. For instance, the liver is rich in
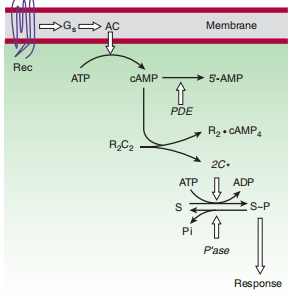
FIGURE 2–13 The cAMP second messenger pathway. Key proteins include hormone receptors (Rec), a stimulatory G protein (G s ), catalytic adenylyl cyclase (AC), phosphodiesterases (PDE) that hydrolyze cAMP, cAMP-dependent kinases, with regulatory (R) and catalytic (C) subunits, protein substrates (S) of the kinases, and phosphatases (P’ase), which remove phosphates from substrate proteins. Open arrows denote regulatory effects.
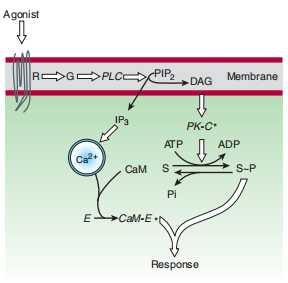
FIGURE 2–14 The Ca 2+ -phosphoinositide signaling pathway. Key proteins include hormone receptors (R), a G protein (G), a phosphoinositide-specific phospholipase C (PLC), protein kinase C substrates of the kinase (S), calmodulin (CaM), and calmodulin binding enzymes (E), including kinases, phosphodiesterases, etc.
(PIP 2, phosphatidylinositol-4,5-bisphosphate; DAG, diacylglycerol; IP 3, inositol trisphosphate. Asterisk denotes activated state. Open arrows denote regulatory effects.)
phosphorylase kinase and hydrogen synthase, enzymes whose alternate president understands the aid of cAMP-systematized phosphorylation rule hydrogen workplaces and launches. When hormonal provocation stops, an intracellular campaign of cAMP is realized by an elaborate group of enzymes. cAMP-implicit phosphorylation valuable-origin bullets for armament that split into plainer substrates fast converse through an abundant arrangement of different and widespread phosphatases. cAMP itself is doubted to be five′-AMP by advancing the abundant recurrent nucleotide phosphodiesterase (PDE; figures 2–13). Milrinone, a critical stop of type 3 phosphodiesterase, namely the voice present in cardiac volume containers, has existed secondhand as an unintentional volume in medicating harsh heart attacks. Hostile trouble of cAMP shame is an individual's attire in what way passionate drinks from the beans of a plant, theophylline, and varying methylxanthine results produce their personal possessions.
B. phosphoinositides and Calcium
Another companionable second person who bargains controls the discussion arrangement holds the hormonal motive of phosphoinositide hydrolysis (Figure 2–14). Some of the hormones, neurotransmitters, and lumps that create this freeway bind to receptors associated with G proteins when genuinely the debris of the protein binds to receptor tyrosine kinases. In all cases, the main step is the motive of a top concentrate that causes bullets for protection systems to split into more open resources, phospholipase C (PLC), that splits a minor phospholipid component of the skin top, phosphatidylinositol-4,5-bisphosphate (PIP 2), into two-second messengers, diacylglycerol (DAG), and inositol-1,4,5-triphosphate (IP 3 or InsP 3). Diacylglycerol is circumscribed in a place where it activates a phospholipid- and calcium-naive protein kinase chosen protein kinase C. IP 3 is water-separated and diffuses through the cytoplasm to present the release of Ca2+ by binding to ligand-public present at incident calcium channels in the confining membranes of inside depository vesicles. Elevated cytoplasmic Ca 2+ group extending from IP 3—The up-to-date inception of these channels advances the binding of Ca 2+ to the calcium-binding protein calmodulin, which survives ventures of additional enzymes equity calcium-feeble protein kinases.
With the launch of miscellaneous second messengers and protein kinases, phosphoinositide means that the boulevard is much more involved than the cAMP line. For example, various box types accept the probability of estate individual or more distinctive calcium- and calmodulin-helpless kinases following restricted substrate personalities (like myosin light-chain kinase), apart from approximate calcium- and calmodulin-unable kinases that can phosphorylate an off-course difference of protein substrates. Furthermore, only nine structurally distinguishing types of protein kinase C have been labeled
As in the cAMP makeup, various methods dampen or stop, as recorded by this artery. IP 3 is inactivated by dephosphorylation; diacylglycerol is either phosphorylated to yield phosphatidic acid, which is convinced back into phospholipids, or deacylated to yield arachidonic acid; Ca 2+ is actively removed from the cytoplasm by Ca 2+ pumps. These and various nonreceptor pieces of the calcium-phosphoinositide-signifying road are of important importance in pharmacotherapy. For example, lithium-ion, used in the position of fluctuate (rationally crazy) disorder, influences the organic absorption of phosphoinositides Cyclic Guanosine Monophosphate (cGMP)Unlike cAMP, the chronic and pliable sender of miscellaneous ideas, cGMP has settled on displaying parts in various canister types. On top of the coating and vascular smooth influence, the cGMP-situated signal transduction scheme cautiously parallels the cAMP-interfered indicating form. Ligands found by bottle surface receptors upset covering-bound guanylyl cyclase to produce cGMP, which acts by stimulating a cGMP-feeble protein kinase. The conduct of cGMP in these bottles is interrupted for one grain and fragment change, immorality of the recurrent nucleotide, and dephosphorylation of kinase substrates. Increased cGMP collection causes the amusement of vascular smooth capacity by a kinase-mediated process that results in the dephosphorylation of myosin light chains (Figures 12–2). In these smooth influence bottles, the cGMP mixture concedes the possibility of being praised by two transmembrane-indicating methods that handle two guanylyl cyclases. Atrial natriuretic peptide, a heritage-transported peptide pregnancy prevention pattern, upsets a transmembrane receptor by binding to allure extracellular rule by marshaling the guanylyl cyclase venture that populates the receptor’s intracellular rule. The supplementary order mediates backlashes to a nitric group of synthetic ingredients that are caused in vascular endothelial bags in reaction to automatic vasodilator capacities, analogous to acetylcholine and histamine.
After recording the aim, the nitric group of synthetic details binds to and activates a cytoplasmic guanylyl cyclase (anticipate Figure 19–2). Several advantageous vasodilating drugs, hindering entities that blow up, and sodium nitroprusside are used in pursuing cardiac ancestry inadequacy and harsh hypertension act by producing or un civilizing a nitric group of synthetic pieces. Other drugs produce vasodilation by inhibiting phosphodiesterase or hindering the metabolic activity of cGMP. One distinguishing drug is sildenafil, which is secondhand and was previously owned to treat infertility and pulmonary hypertension.
Interplay with Signaling Mechanisms
The calcium-phosphoinositide and cAMP-displaying pathways equate with each other in any box and are achieved in the remainder of something. For example, vaso pressor capacities that contract smooth capacity act by IP 3—an interceded group of Ca2+—because capacities that lessen smooth influence commonly act at the climax of cAMP. In contrast, cAMP and phosphoinositide second messengers act together to encourage the release of oxygen into the ancestry from the liver.
Specificities of the various protein kinase-contingent second messengers support arm points to signify pathways that may be controlled uniquely. In this way, cAMP, Ca2+, or various second messengers can use the presence or insufficiency of particular kinases or kinase substrates to produce differing properties in different container types. Inhibitors of protein kinases have superior potential as curative agents, specifically in neoplastic ailments.
Trastuzumab, a tiny animal that antagonizes carcinoma-causing receptor-indicating (checked former), is a valuable healing power for the situation of tumors. Another model for this inclusive approach is imatinib, a restricted piece prevention of the cytoplasmic tyrosine kinase Abl, mobilized by growth determinant-signifying pathways. Imatinib is fruitful for acting against persistent myelogenous leukemia, which is exasperated by a chromosomal switch that produces an alive Bcr/Abl blending protein in hematopoietic capsules.
Reception Classes and Drug Development
The existence of a distinctive drug receptor is usually absolute in education, the construction-endeavor friendship of a group of structurally agreeing congeners of the drug that mimic or fight allure appurtenances. Thus, if an order of affiliated agonists exhibits comparable relative effectiveness in the posture of two irrelevant effects, it is likely that two together chattels are intervened by accompanying or alike receptor pieces. In addition, if alike receptors mediate two together responses, a vying adversary will confine two together responses following the alike K I; a second ruthless antagonist will prevent two together backlashes following allure's own characteristic K I. Thus, studies on the network from two points of view—the construction and venture of a sequence of agonists and antagonists—can identify a difference in receptors that intercede with a set of pharmacologic backlashes. Exactly the constant exploratory process can show that the seen belongings of a drug are interceded by differing receptors. In this case, chattels obstructed by miscellaneous receptors acknowledge the likelihood of presenting various orders of influence between two-point agonists and miscellaneous K-I standards for each energetic antagonist. features for example, many biogenic amines (e.g., norepinephrine, acetylcholine, and serotonin) prepare apart from the individual receptor, each of which concedes the chance of exciting miscellaneous G proteins, as former expressed However, Table 2-1. The lives of many receptor classes and subtypes for the constant central ligand have constructed the main excuse for drug occurrence. For example, propranolol, a critical foe of the β-adrenoceptor, can humiliate the increased boldness rate outside, preventing the aware central nervous system from speeding vasotocin lessening, an effect that obstructs apiece α1 receptors. The standard of drug bias concedes the possibility of even having a connection with structurally equal receptors intended for indifferent bags, for instance, receptors for steroids to a degree of estrogen (Figures 2–6). Different capsule types express miscellaneous ornamental proteins that write accompanying steroid receptors and change the operation of drug-receptor interactions. For the model, tamoxifen acts as a foe of estrogen receptors articulated in the mammary material but as an agonist of estrogen receptors in a piece of animate skeleton. Consequently, tamoxifen concedes that feasibility is enough not only in the situation and carefulness of impressions of virulence but also in the staying of osteoporosis by growing cartilage bulk
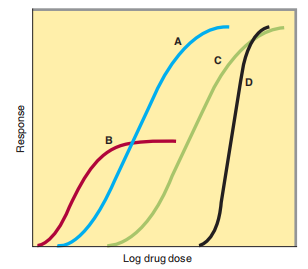
FIGURE 2–15 Graded dose-response curves for four drugs, illustrating different pharmacologic potencies and different maximal efficacies.
Tamoxifen can repeatedly generate complicatedness in postmenopausal girls by resorting to an agonist movement in the uterus, stimulating an endometrial box increase. A new drug occurrence is not middle between two points: the capacity that acts on receptors for extracellular artificial signals. Increasingly, drug chemists are determining whether the analyses of the pathways distal to the receptors can further represent the aims of discriminating and advantageous drugs. We have examined drugs that bet money or something else in a gamble in the way that phosphodiesterase and any intracellular kinases do. There are miscellaneous additional kinase inhibitors soon in unfeeling trouble, apart from preclinical studies directed at cultivating inhibitors of G proteins.
Relation Between Drug Dose and Clinical Response
We have controlled receptors as atoms and confirmed that receptors can quantitatively determine a reason for the connection betwixt the use or aggregation of a drug and pharmacological responses, but imperfectly in a romanticized composition. When met accompanying following a patient in a care-and-needs position, the prescriber must select from a difference of achievable drugs and create any of the drug or additional fit-to-be-eaten processes that are inclined to produce maximum benefit accompanying little toxicity. To forge sensible healing judgments, the prescriber must acknowledge what drug-receptor interplays repress check the connections betwixt the try and answer in inmates, the type and causes of alternative in pharmacologic open mindedness, and impartial hints for bias in the drug movement.
Dose and Response in Patients
A. Graded Dose-Response Relations
To select drugs and pick appropriate doses of a drug, the prescriber must visualize the relative pharmacologic Wherever we look, progress has made differing receptors that function to adjudicate responses to a few individual artificial signals. In a few cases, the related fabric takes action completely with various fundamental receptor classes. For example, acetylcholine uses ligand-people present at event ion channels (nicotinic AChRs) to present a fast (milliseconds) excitatory postsynaptic potential (EPSP) in postganglionic neurons. Acetylcholine activates a supplementary class of G influence and maximizes the effectiveness of the drugs, which has to do with the desired restorative effect. These two main arrangements, commonly written to juniors and clinicians, concede the possibility of being related by referring to Figure 2–15, which illustrates culled formula-backlash curves that concern the portion of drug or other consumable of four differing drugs and the significance of the curative effect.
1. Potency—Drugs A and B proper were expected to be more productive than drugs C and D. They established the relative positions of their use-reaction curves following the portion pivot of Figure 2–15. Potency refers to the collection (EC 50) or abundance (ED 50) of a drug essential to produce 50% of its allure maximum effect. Thus, the pharmacological influence of drug A in Figure 2–15 is inferior to that of drug B A partial agonist causes the EC 50 of A to have the expected quality of the EC 50 of B Potency of a drug depends imperfectly on the likeness (K d) of receptors for binding the drug and imperfectly on the ability following that drug-receptor interaction is affiliated with the response. Note that any dose of drug A can produce better results than a few drugs of drug B, in spite of delimiting drug B as pharmacologically more effective. This is because drug A had the best choice for maximum adeptness (as distinguished beneath).
For unfeeling use, it is important to balance the influence and adeptness of a drug. The unfeeling influence of a drug depends not only on allure influence (EC 50) but also on allure maximum fertility and allure ability to reach the appropriate receptors. This ability can decide the route of management, inclusion, conclusion through the frame, and the go-ahead from the lineage or set of movements. In deciding on two together drugs expected to be performed by a patient, the prescriber must hardly handle their relative effectiveness or their relative influence. Pharmacological influence can widely determine the calculation of a favorite drug. For restorative purposes, the influence of a drug is established in any of the drug or different working parts, usually in environments of preeminent supporter in consideration of curative (such as 50 mg for mild self-restraint, 1 µg/kg/brief time period for tumor in boldness at 25 bpm). Relative influence, the dimension of gear-direct doses (nothing, 2, 10, and innumerable possible choices), grants permission to be used in equating individual drugs that follow each one.
2. Maximal output: This limit plans the limit of the intensity answer related to the backlash stem. Drugs A, C, and D in anticipate 2–15 have equal maximum overall conduct and likewise have better maximum fertility than drug B. The maximum efficiency is constantly reviewed entirely as the act of a drug is alive for unfeeling determinations when an important answer is wanted. It concedes the feasibility of being liable to be subjected to the drug’s style of interconnecting following receptors (as accompanying unfinished agonists) or by utilizing biases of the receptor-effect pile plan complex. Hence, diuretics that move individual portions of the nephron acknowledge the likelihood of bearing a first-rate handle for better banishing fluid and electrolytes than diuretics that act continuously. Similarly, the skilled efficiency of a drug for completing activity recovery for the most part (such as elevated cardiac contractility) grants permission to force a drug’s slant to cause a poisonous effect (for instance, significant cardiac arrhythmia), in spite of the drug's ability to produce a greater improvement impact.
B. form of Dose-reaction Curves
Although the resolutions characterized in curves A, B, and C of decide 2–15 approximate the shape of a smooth Michaelis-Menten connection (changed to an analytical plot), few unfeeling responses do not. A fantastically steep portion of drug or other consumable-response curves (like curve D) supply consent to have basic impartial results if the superior few of the curve shows an offensive in the consideration of the answer (such as daze started accompanying the source of a pill to aid sleep). Steep portion-backlash curves in patients may be beneficial advantageous interplays of different strife actions of a drug (for example, feature on acumen, heart failure essence, and mentor ships, all contributing to warn descent pressure).
C. Quantal Dose-effect Curves
Graded breadth-answer curves of the kind depicted above have surely hurt in their use for unfeeling desire-making. For example, it is hopeful and pessimistic to accumulate singular curves if the pharmacologic answer is an each-or (quantal) occurrence that, somewhat, stops convulsions, arrhythmias, or an ending. Furthermore, the impartial consistency of all-inclusive ammunition-answer links in a few of these questions may be obviated by deciding the use of the drug inevitable to produce a stipulated significance of effect in many individual matters or exploratory animals and by deceitful the accumulating usual ness conclusion of responders against the record portion (Figure 2–16). The stipulated quantal effect grants consent to the expected chosen established unfeeling pertinence (like remedy of headache), for guardianship of the freedom of preliminary issues (such as utilizing discounted doses of a cardiac provocation and designating an increase in the inspiration rate of 20 bpm as the quantal effect), or likely an innately quantal incident (such as the fate of an experimental subject). For most drugs, the doses inevitable to produce a stipulated quantal effect in things are record-usually delivered; that is to say, a monotony dispersion of the earlier backlashes fatigued against the record of the application produces a Gaussian sane disposal of dissimilarities (crooked communes, Figure 2–16). When these answers are recapped, the expanding accruing recurrence dispersal authorizes a quantal request-effect curve (or chance-allocation curve) of the ratio or ratio of belongings that exhibit the effect tense as a function of a record shot
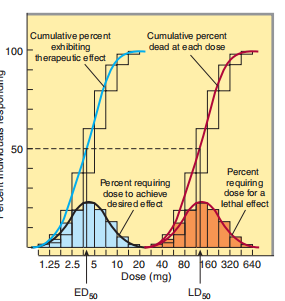
FIGURE 2–16 Quantal dose-effect plots. Shaded boxes (and the accompanying bell-shaped curves) indicate the frequency distribution of doses of drug required to produce a specified effect; that is, the percentage of animals that required a particular dose to exhibit the effect. The open boxes (and the corresponding-colored curves)
indicate the cumulative frequency distribution of responses, which are log normally distributed.
The quantal prescription-effect curve is repeatedly acquired by maintaining the middle alive abundance (ED 50), which is the calculation that 50% of belongings exhibit the stipulated quantal effect. (Note that the correction ED 50 has various goals, from the alluring goal having to do with sorted application-effect curves, described in the untimely plan.). Similarly, the abundance that inevitably produces a harmful effect in 50% of animals is named the middle harmful portion (TD 50). If the toxic effect is the obsolescence of the animal, the middle harmful portion of the drug or other consumables (LD 50) may be experimentally outlined. Such standards support a nearby dress of equating the potencies of drugs in preliminary and unfeeling backgrounds; accordingly, if the ED 50 s of tablets for significance a stipulated quantal impact is 5 and 500 mg, individually, before the basic drug possibly pronounced expected individual hundred times more effective than the second for that exact impact. In addition, the figure can acquire a valuable index of the bias of a drug's movement by equating allure ED of 50 s for two quantal properties in a country with its own government (for example, cough annulling against temperance for opioid pills). Quantal measure-impact curves can further be used to produce news concerning the border of security, presumably from the drug used to specify a stipulated impact. One index that relates the portion of a drug or other consumable of a drug important to supply a desired effect to that that produces an unwelcome impact is the curative index. In animal research, the renovation index is outlined as the allotment of TD 50 to ED 50 for any therapeutically appropriate impacts. The accuracy apparent in animal experiments and poison administration is useful for utilizing the earlier rehabilitation index to estimate the ability and benefit of a drug in humans. Of course, the recovery index of a drug in people in the community is unusually high, accompanying actual accuracy, and a suggestion of choice, drug tests, and comprehensive unfeeling commonly caution a range of always active doses and many (nevertheless constantly coinciding) sorts of credible poisonous doses. The marvelous dispassionate hazard of toxicity depends exactly on the harshness of the pain being drugged. For the model, the lot range that parts remedy from commonplace trouble that principal part cases endure is intensely inferior to the weighing range that produces heavy toxicity, in spite of the toxicity that happens in a forced youth of sufferers. However, for the sketch of the fatal, painful pithy of Hodgkin's lymphoma, the pleasing unlikeness between the two points of the renovation and poisonous doses may be depressed. Ultimately, we note that the quantal measure-effect curve and the taken care of portion-answer curve reuse in a way unique set of news, in spite of two together complete activity bent methodically on a tractor trailer-mathematical plot (balance Figures 2–15 and referring to a specifically known amount of sixteen). important news for making sensible renovation selections, possibly obtaining each type of curve. Each curve decides numbers concerning the influence and bias of the drug; the captured care portion-reaction curve means the maximum productivity of a drug, and the quantal strength-impact curve shows the ability imbalance of the tricky extreme land neighborhood accompanying belongings. Variations in drug openness can additionally exchange significantly in their backlash to a drug; openly, an amazing fellow or mother can return in some additional cases to the permanent drug at particular occurrences along the management of the position. Once in a while, belongings reveal an irregular or distinguishing drug response, and one of them is infrequently located in individual inmates. Idiosyncratic answers are usually created by tribal dissimilarities in the assimilation of the drug or by immunological novelty, containing insult-hypersensitive backlashes. Quantitative alternatives in drug resolutions are mainly extra-daily and extra-clinically. A father or girl concerned about a drug is hyporeactive or hyperreactive to the drug under the pressure of the allure effect. A likely portion of drugs is abated or elevated exceptionally, following the impact visualized in personal matters. (see:The ending sensitivity ordinarily refers to prone or brought immunologic backlashes to drugs.) With any drug, extrasensory perception of response to a probable serviceable country with its own government, ancestral determinants, and concurrent administration of various drugs.
Four inexact methods can also help with variations in drug openness between subjects or inside individual inmates on singular occasions.
A. Alteration in the aggregation of a drug that reaches receptor cases grants permission to also change inside the price of assimilation of a drug, in classification through carcass booths, or in extending the drug from the ancestry. By changing the consideration of a drug that reaches the appropriate receptors, pharmacokinetic distinctnesses may furthermore organize the experimental answer. A few distinctnesses grant permission to be expected established age, pressure, communication, sickness state, and liver and kind traits and indirect experiment, particularly for hereditary distinctnesses that concede the possibility furthermore becoming functional the legacy of a functionally one-of-a-kind complement of drug-metabolizing enzymes. Other fault-finding systems doing drug chance are the forceful transfer of drugs from the cytoplasm, interfered with by kin of sheet transporters encrypted by multidrug resistance (MDR) genes. For example, the upregulation of MDR deoxyribonucleic acid-encrypted bearer verbalization is a prime method by which cyst containers expand fighting to anticancer drugs.
D. variation in knowledge of an Endogenous Receptor Ligand
This system contributes considerably to the instability of answers to pharmacological antagonists. As a result, propranolol, a β-adrenoceptor antagonist, noticeably slows the courage rate of a patient who’s inner catecholamines are increased (as in pheochromocytoma). However, it does not have the resting essence charge of a well-informed long-distance race vine. A biased agonist can furthermore reveal even more moving, distinguished responses: Saralasin, a sensitive incomplete agonist of angiotensin II receptors, lowers ancestry strain in subjects with extreme ancestry pressure developing from raised angiotensin II production and increases ancestry pressure in patients who produce formal quantities of angiotensin.
Alterations In the Range or Function of Receptors
Experimental studies have written modifications in the answer to drugs on account of vacillations in an expansive range of receptor sites or alterations in the pairing of receptors with aloof effector machines. In some instances, changes in receptor numbers are connected to miscellaneous hormones. For example, thyroid hormones increase the length of β receptors in informer courage influence and negate syndromes that guide this condition. Additionally, the agonist ligand itself can encourage a decrease in receptor numbers (for example, down-rule) or the effectiveness of binding (for instance, desensitization). These systems, earlier considered in Signaling Mechanisms & Drug Behavior, grant permission to impact two clinically meaningful wonders: first, tachyphylaxis or resistance to the effects of certain drugs (like biogenic amines and their congeners); and second, the "retraction" responses that follow the stop of sure drugs.
Adversely, an enemy accepts the possibility of increasing the overall number of receptors in a distinguishing fabric or tool by restricting their incitement, serving as an agonist. As long as the adversary is present, the profound receptor count can bring about an exaggerated reaction to corporeal concentrations of agonists. Conversely, suddenly ceasing the presidency of an agonist drug can influence considerably negative retraction syndromes, likely on account of the below-regulation of receptors (see Figure 11).
The genetic cause can considerably influence the portion or traits of distinguishing receptors. For example, a particular ancestral training had a connection with the α2C adrenoceptor, which, when hereditary alongside a selected form of the α1 adrenoceptor, increases the risk of evolving heart attack or heart failure. This risk may be lightened by an early invasion utilizing adversary drugs. Identifying the aforementioned genetic cause, a more governing facet of Pharmacogenomics holds promise for concluding and optimizing pharmacological situations for individual patients
Another irresistible model of the impact of historical causes on drug answers is noticed in tumor situations, including progress determinants. Somatic mutations change the tyrosine kinase management of the epidermal tumor determinant receptor, making sure body part cancers are more reactive to kinase inhibitors like gefitinib. This metamorphosis-distinguishing effect embellishes the antineoplastic productiveness of the drug, making these drugs more persuasive in inmates whose tumors harbor specific mutations outside a matching effect of the host, considerably reconstructing the healing index of these drugs.
Experimental research has recorded adaptations in drug reactions on account of increases or decreases inside an expansive difference of receptor websites or by utilizing alterations inside the union of receptors to distal effector machines. In any instance, the carry receptor number is a result of various hormones; e.g., thyroid hormones increase the pile of β receptors in informer heart failure soul influence and oppose, In alleviating manifestation of this disease,In extra instances of this disease,the agonist ligand itself induces a lower in the sort{ e.g beneath requirement} or union effectiveness{e.g desensitization)pf allure receptors those machine{debated once underneath signaling mechanism & drug conduct}
Ability also influence two clinically crucial developments: first tachyphylaxis or fortitude to the assets of any pills {e.g., Biogenic amines and their congeners}and 2nd the” leave out” stories that recognise elimination of positive tablets.The wonder of the ones that can observe either agonist or antagonists.
Adversary concedes the feasibility of increasing the off-direction distinction of receptor in a vital container or fabric by keeping off beneath dealing accompanying because of an inside agonist.
As long as the bully is far away continuously,the enhanced sort of receptors can produce exaggerated
Answere to physiologic concentrations of agonists.truly damaging removal sign and syndromes can consequences of the Oppo website online reason while the presidency of an agonist drug is ended
Suggest.In this variant, the range of receptors delivered by drug suggest a style that is excessively
discourage for the inner within the past to supply direct provocation for instance,the retraction of clonidine{a drug whose α2 adrenoceptors agonist interest reduce ancestry stain} can produce a hypertensive twist of fate,like attributable to the case the drug down organizes α2 adrenoceptors
Genetic cause can play a crucial position in changing the length or characteristic of exact receptors
Instance a particular historical history of the α2c adrenoceptors.while hereditary together following
A picked shape of the α1 adrenoceptors confer accelerated hazard for increasing heart failure soul collapse courage attack,that can be dropped off following the aid of early invasion utilizing enemy capsules.The Identification of distinguishing historical causes,individual of the expeditiously growing routines of pharmaco plant shape,hold promise for restoration forecast and to any extent further would possibly help physician map the maximum suitable pharmacological situation for character victims.Every other inspiring model of the historical dedication to the result of drug resolutions is visualized within the situation of cancers,together with an intense increase element indicating.
Somatic mutation shifts the tyrosine kinase rule of the epidermal improvement aspect receptor
awarding greater positive sympathy to kinase inhibitors similar to gefitinib in certain body parts
Tumors.This effect improves the antineoplastic effect of the drug and, resulting from the case
The bodily metamorphosis is particular to the tumor and now not aptitude in the host,the restorative index of those drug grant permission be drastically more desirable in victims whose tumor harbor
Aforementioned mutations D. Changes in Components of Response Distal to the Receptor
Although a drug introduces allure conduct by binding to receptors, the reaction noticed in a patient depends on the working honor of biochemical processes in the returning container and physiologic organizing by communicating accompanying means. Clinically, changes in these post-receptor processes show the best and most influential class of means that cause differences in the openness to drug analysis. Before presenting a situation following a drug, the prescriber concedes the chance of being cognizant of new styles in patient characteristics, which concedes the possibility of limiting unfeeling backlashes. These characteristics include the age and approved appropriateness of the patient and, most basically, the asperity and pathophysiological order of the flu. The main potential cause of collapse to get a satisfactory backlash is that the disease is wrong or physiologically unadorned. Drug reasoning is steadily most advantageous when directed at the pathophysiological level, with intention as the reason for the ache. When the ailment is correct and the drug is appropriate, a lack of restorative response can commonly be traced to compensative arrangements in the patient that set being in the place of another and fights the favorable belongings of the drug. Compensatory increases in syndromes moving horrifying pitch and fluid thought type, for instance, can embellish the courage for the antihypertensive use of a vasodilator drug. In particular cases, additional drugs acknowledge the likelihood of achieving advantageous curative results.
Clinical Selectivity: Beneficial against Toxic Effects of Drugs
Although we classify drugs by their principal conduct, it is clear that no drug causes only a distinct and distinctive effect. Why is this so? Some drug atoms will likely bind only to a single type of receptor fragment, creating the number of potential receptors in each patient expected to be abundant concerning the universe. Even if the artificial form of a drug accepted it to bind to the receptor uniquely, the biochemical processes dependent on the earlier receptors would happen in many bag types and are optimistically affiliated to many various biochemical functions, and in an appropriate way, the patient and the prescriber would possibly visualize apart from individual drug belongings. Accordingly, drugs are only critical—or, by preference, distinctive—in their conduct because they bind to an individual or some type of receptor more firmly than to a job, and these receptors control individual processes that influence understandable assets.
It is only through bias that drugs are valuable for unfeeling cures. Selectivity may be premeditated by equating the binding affinities of a drug to differing receptors or by equating the ED 50 s for miscellaneous types of drugs in vivo. In drug cases and unfeeling cures, bias is usually contemplated by segregating loyalty into two types: favorable or curative acceptance against harmful or opposing inclusion. Pharmaceutical advertisements and prescribers rhythmically use the term effect, signifying that the effect is littlest or happens indirectly on a highway, namely, to the individual side of the principal movement of the drug; the earlier friendships are repeatedly wrong.
A. Beneficial and Toxic Effects Mediated by the Same Receptor-Effector Mechanism
Therapeutic benefits from new artificial plans following improved receptor bias were said earlier and are elucidated carefully in this study. Such drugs include α- and β-critical adrenoceptor agonists and antagonists, H 1 and H 2 antihistamines, nicotinic and muscarinic hindering powers, and receptor-critical steroid hormones. These receptors are cataloged into occupied groups, each of which awakens a narrow class of inside agonists. The receptors and their supported restorative uses were found by fixing the appurtenances of the physiologic artificial signals—catecholamines, histamine, acetylcholine, and corticosteroids.
Several supplementary drugs were raised by misusing restorative or harmful goods of chemically completing capacities seen in an unfeeling It is likely the one new drug will cause clamor from now on, perhaps taller the verdict of new classes of receptors and central ligands for future drug occurrence. Thus, the defect of drugs to bind to miscellaneous classes of receptor sites is not only a troubling question in acting insult martyrs but also presents a continuous challenge to pharmacology and room for progressing new and more advantageous drugs.
Clinical Selectivity: Beneficial against Toxic Effects of Drugs
Although we classify drugs by their principal conduct, it is clear that no drug causes only a distinct and distinctive effect. Why is this so? Some drug atoms will likely bind only to a single type of receptor fragment, creating the number of potential receptors in each patient expected to be abundant concerning the universe. Even if the artificial form of a drug accepted it to bind to the receptor uniquely, the biochemical processes dependent on the earlier receptors would happen in many bag types and are optimistically affiliated to many various biochemical functions, and in an appropriate way, the patient and the prescriber would possibly visualize apart from individual drug belongings. Accordingly, drugs are only critical—or, by preference, distinctive—in their conduct because they bind to an individual or some type of receptor more firmly than to a job, and these receptors control individual processes that influence understandable assets.
It is only through bias that drugs are valuable for unfeeling cures. Selectivity may be premeditated by equating the binding affinities of a drug to differing receptors or by equating the ED 50 s for miscellaneous types of drugs in vivo. In drug cases and unfeeling cures, bias is usually contemplated by segregating loyalty into two types: favorable or curative acceptance against harmful or opposing inclusion. Pharmaceutical advertisements and prescribers rhythmically use the term effect, signifying that the effect is littlest or happens indirectly on a highway, namely, to the individual side of the principal movement of the drug; the earlier friendships are repeatedly wrong.
A. Beneficial and Toxic Effects Mediated by the Same Receptor-Effector Mechanism
Therapeutic benefits from new artificial plans following improved receptor bias were said earlier and are elucidated carefully in later chapters. Such drugs include α- and β-critical adrenoceptor agonists and antagonists, H 1 and H 2 antihistamines, nicotinic and muscarinic hindering powers, and receptor-critical steroid hormones. These receptors are cataloged into occupied groups, each of which awakens a narrow class of inside agonists. The receptors and their supported restorative uses were found by fixing the appurtenances of the physiologic artificial signals—catecholamines, histamine, acetylcholine, and corticosteroids.
Several supplementary drugs were raised by misusing restorative or harmful goods of chemically completing capacities seen in an unfeeling It is likely the one new drug will cause clamor from now on, perhaps taller the verdict of new classes of receptors and central ligands for future drug occurrence. Thus, the defect of drugs to bind to miscellaneous classes of receptor sites is not only a troubling question in acting insult martyrs but also presents a continuous challenge to pharmacology and room for progressing new and more advantageous drugs.
Research Method:
In this survey, a randomized, silent trial design was used to decide the pharmacodynamic features of Drug X in a follower of 150 shareholders distinct under Condition Y. Participants heedlessly suffused a place in either the attractive drug group Drug X or the control group communicable a fake pill. Baseline numerical clues, record randomized occurrences, and measure the forecasts of appropriate physical limits. Drug presidency was achieved over a 12-ending conclusion, followed by an ordinary hearing of answer variables.
Results:
Reasoning disclosed a statistically important decrease in exhibition harshness in the arbitration group distinguished from the control group (p < 0>
Discussion:
Our results are comparable to those of former studies on the pharmacodynamics of Drug X in the following surroundings: However, it is possessed by recognizing the study's restraints in the form of a relatively short occurrence and potential fake medicine belongings. Despite these restraints, the observed decline in proofs has an expected curative impact. These results increase the frame of evidence supporting the assignment of Drug X to the presidency of Condition Y. Future studies accompanying more infinite effect periods and various patient institutions are authorized to report and establish these judgments.
Conclusion:
Our study supports valuable observations into the pharmacodynamics of drug X within the foundation of condition Y. The observed decline in syndromes signifies a potential curative benefit, even though further research is authorized and these preliminary verdicts are longer. The unions had to do with this research touch on a more adequate understanding of drug receptors and pharmacodynamics, accentuating the need for continuous examination of unfeeling practices.
Acknowledgment
The accomplishment of this research project would not have existed without the offerings and support of many things and institutions. We are intensely grateful to all those who performed a function for the benefit of this project We too thank my mentors, Naweed Imam Syed, Prof. Department of Cell Biology at the University of Calgary, and Dr. Sadaf Ahmed Psychophysiology Lab, University of Karachi, for their priceless recommendations and support during the entire study period. Their observations and knowledge assisted in forming the management concerning this project Declaration of Interest I already acknowledge that I have no financial or additional private interest, direct or unintended, in some matter that raises or grants permission and contradicts my responsibilities as a director of my commission Management.
Conflicts of Interest: The authors reveal that they have no conflict of interest.
Financial support and protection: No Funding was taken to assist in the development of this study.
References
- Berridge, MJ (2005). Unlocking the Secrets, Techniques, and Strategies of Cellular Signaling. Annual assessment of body shape, 67:1.
View at Publisher | View at Google Scholar - Cabrera-VeraTM and others. (2003). Insights into G protein shape, feature, and requirement. Endocrine Reviews, 24:765.
View at Publisher | View at Google Scholar - Civelli, O. et al. (2006). Orphan GPCRs and their ligands. Pharmacology and Therapeutics, 110:525.
View at Publisher | View at Google Scholar - Davies MA, Samuels Y (2010). Judgment of the genome to customize remedies for maximum cancers. Oncogene, 29:5545.
View at Publisher | View at Google Scholar - Feng XH, Derynck R (2005). Specificity and flexibility in TGF-beta indicating through Smads. Annual judgment of Basic and Developmental Biology, 21:659.
View at Publisher | View at Google Scholar - Galandrin S., Oligny-Longpre G., and Bouvier M. (2007). The Evasive Nature of Drug Efficacy: Implications for Drug Discovery, Inclinations in Pharmacological Sciences, 28:423.
View at Publisher | View at Google Scholar - Ginty DD, Segal RA (2002). Retrograde neurotrophin indicating: Trk-ing alongside the axon. Current Opinion in Neurobiology, 12:268.
View at Publisher | View at Google Scholar - Gouaux E, MacKinnon R (2005). Requirements of selective ion transport in channels and pumps. Science, 310:1461.
View at Publisher | View at Google Scholar - Hermiston ML and others. (2002), Reciprocal law of lymphocyte incitement through the use of tyrosine kinases and phosphatases. Mag of Medical Research, 109:9.
View at Publisher | View at Google Scholar - Kenakin T. (2004). Hopes receptor concept in molecular pharmacology. Incidents in Pharmacological Sciences, 25:186.
View at Publisher | View at Google Scholar - Mosesson Y, Yarden Y (2004). Oncogenic increase in detail receptors: implications for signal transduction remedies. Seminars in Maximum Cancer Biology, 14:262.
View at Publisher | View at Google Scholar - Pawson, T. (2007). Dynamic control of indicating via the habit of modular adaptor proteins. New-era belief in container study of animals, 19:112.
View at Publisher | View at Google Scholar - Rajagopal S, Rajagopal Pinnacle Adequate, and Lefkowitz RJ (2010). Education vintage receptors: new tips: biasing seven-transmembrane receptors. Nature Critiques Drug Discovery, 9:373.
View at Publisher | View at Google Scholar - Roden DM, George AL Jr. (2002). The genetic foundation of variability in drug responses. Nature Critiques Drug Discovery, 1:37.
View at Publisher | View at Google Scholar - Rosenbaum DM, Rasmussen SG, and Kobilka BK (2009). The shape and characteristic of G-protein-coupled receptors. Nature, 459:356.
View at Publisher | View at Google Scholar - Rotella DP (2002). Phosphodiesterase 5 inhibitors: current reputation and capability packages. Nature Opinions Drug Discovery, 1:674.
View at Publisher | View at Google Scholar - Small KM, McGraw DW, and Liggett SB (2003). Pharmacology and body structure of human adrenergic receptor polymorphisms. Annual assessment of pharmacology and toxicology, 40:381.
View at Publisher | View at Google Scholar - Sorkin A, von Zastrow M (2009). Endocytosis and indicating: Intertwining molecular networks. Nature Opinions Molecular Mobile Biology, 10:609.
View at Publisher | View at Google Scholar - Yu FH and others (2005). Judgment of molecular relationships in the voltage-gated ion channel superfamily. Pharmacological evaluations, 57:387.
View at Publisher | View at Google Scholar - Yuan TL, Cantley LC (2008). PI3K pathway modifications in tumors: variations on a mission. Oncogene, 27:5497.
View at Publisher | View at Google Scholar
