

Review Article | DOI: https://doi.org/10.31579/2834-8761/006
What The Body Does to A Drug: Pharmacokinetics
- Gudisa Bereda 1*
Department of Pharmacy, Negelle Health Science College, Guji, Ethiopia
*Corresponding Author: Gudisa Bereda, Department of Pharmacy, Negelle Health Science College, Guji, Ethiopia.
Citation: Gudisa Bereda. (2022). What The Body Does To A Drug: Pharmacokinetics, J.Clinical Endocrinology and Metabolism, 1(1) DOI: 10.31579/2834-8761/006
Copyright: © 2022 Bon E.I Gudisa Bereda. This is an open-access article distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Received: 06 September 2022 | Accepted: 23 September 2022 | Published: 24 October 2022
Keywords: body; drugs; pharmacokinetics
Abstract
Pharmacokinetics may be defined as the study of the dynamic movements of foreign chemicals (xenobiotics) during their passage through the body and as such encompass the kinetics of absorption, distribution, biotransformation/metabolism and excretion. Absorption is the process that brings a drug from the administration, e.g., tablet, capsule, into the systemic circulation. Bioavailability is the fraction of the originally administered drug that arrives in systemic circulation and depends on the properties of the substance and the mode of administration. It can be a direct reflection of medication absorption. Distribution describes how a substance is spread throughout the body. This varies based on the biochemical properties of the drug as well as the physiology of the individual taking that medication. In the body, a drug may be protein-bound or free. Only free drug can act at its pharmacologically active sites, e.g., receptors, cross into other fluid compartments, or be eliminated. Metabolism is the processing of the drug by the body into subsequent compounds. Excretion is the process by which the drug is eliminated from the body. The pharmacokinetic term half-life (t1/2) refers to the time taken for half the initial dose of medicine administered to be eliminated from the body. After three to five half-lives the drug is considered undetectable and unable to exert a pharmacodynamic effect.
Introduction
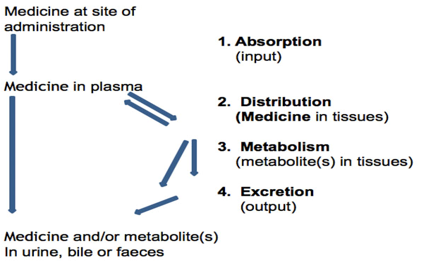
Pharmacokinetics is derived from ancient Greek. Pharmakon means 'drug' and kinetikos means ‘moving or putting in motion [1]. Pharmacokinetics, sometimes described as what the body does to a drug, refers to the movement of drug into, through and out of the body the time course of its absorption, bioavailability, distribution, metabolism, and excretion. Pharmacokinetics (PK) is the study of a drug and/ or its metabolite kinetics in the body. It refers to the temporary evolution of a drug and its metabolites in serum, plasma, or whole blood, tissue target and target organs over time. Another easy way to remember what pharmacokinetics means is to reference the definition of ‘kinetics’. Kinetics essentially means movement, but by definition, it is the study of forces acting on mechanisms. The body is a very complex system and a drug undergoes many steps as it is being absorbed, distributed through the body, metabolised, and/ or excreted (ADME) [2]. Pharmacokinetics may be defined as the study of the dynamic movements of foreign chemicals (xenobiotics) during their passage through the body and as such encompass the kinetics of absorption, distribution, biotransformation/metabolism and excretion (ADME). It can simply be described as how the body handles xenobiotics [3-5].
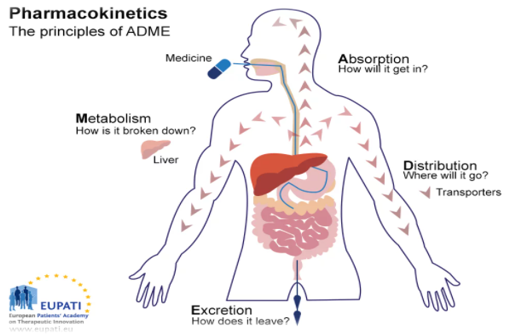
Pharmacokinetics of a drug depends on patient-related factors as well as on the drug’s chemical properties. Some patient-related factors (eg, renal function, genetic makeup, sex, age) can be used to predict the pharmacokinetic parameters in populations. For example, the half-life of some drugs, especially those that require both metabolism and excretion, may be remarkably long in older people. In fact, physiologic changes with aging affect many aspects of pharmacokinetics. Other factors are related to individual physiology. The effects of some individual factors (eg, renal failure, obesity, hepatic failure, dehydration) can be reasonably predicted, but other factors are idiosyncratic and thus have unpredictable effects. Because of individual differences, drug administration must be based on each patient’s needs traditionally, by empirically adjusting dosage until the therapeutic objective is met. This approach is frequently inadequate because it can delay optimal response or result in adverse effects [6].Application of pharmacokinetic principles to individualize pharmacotherapy is termed therapeutic drug monitoring. The type of response of an individual to a particular drug depends on the inherent pharmacological properties of the drug at its site of action. However, the speed of onset, the intensity and the duration of the response usually depend on parameters such as: the rate and extent of uptake of the drug from its site of administration; the rate and extent of distribution of the drug to different tissues, including the site of action; the rate of elimination of the drug from the body [7, 8]. Pharmacokinetics influences the decided route of administration for a specific medication, the amount and frequency of each dose and its dosing intervals [9]. Potential drugs need appropriate pharmacokinetic properties to become safe, useable, effective therapeutics. In order to have a ‘good’ pharmacokinetic profile, a drug must: get into the bloodstream (A); move to the site of action (D); remain unchanged long enough to have a therapeutic effect and then be converted to safe metabolites (M); be adequately cleared (E) [10, 11]. Pharmacokinetics has been broadly divided into two categories of study: absorption and disposition. Disposition means the movement of medicines from site of application to different body fluids. Disposition is further subdivided into the study of distribution and elimination. The term elimination includes metabolism and excretion [12]
Absorption
Absorption is the process that brings a drug from the administration, e.g., tablet, capsule, into the systemic circulation. Absorption affects the speed and concentration at which a drug may arrive at its desired location of effect, e.g., plasma. The process of absorption also often includes liberation, or the process by which the drug is released from its pharmaceutical dosage form [13]. Medicine-related factors include ionisation state, molecular weight, solubility and formulation. Small, nonionised, lipid-soluble medicines permeate plasma membranes most readily. Small molecules typically traverse membranes throughout this process, sometimes via passive transport, but often by way of proteins known as drug transporters. Drug transport can be a critical component of a drug’s disposition in many steps of the pharmacokinetic journey, and preclinical studies should be conducted to provide information on how a drug interacts with various transporters as either substrates or inhibitors [14]. Absorption depends on the administration route and can either be enteral (by in the GI tract, such as by mouth, feeding tube, or rectal suppository) or parenteral (not in the GI tract, such as an injection or topical medication). Additional factors that can affect the amount of the drug absorbed include the drug's formulation (extended release versus immediate release), blood flow to the area of absorption, and GI motility (for enteral medications). The rate and extent of drug absorption depend on multiple factors, such as: route of administration; the formulation and chemical properties of a drug; drug-food interactions. X-rays decrease drug absorption by forming vasoconstriction while parasympathetic increases drug absorption by relaxing the smooth muscle of blood vessels. Gastric emptying time is slower in females’ than males, mainly secondary to the effects of estrogen [15]. The administration (e.g., oral, intravenous, and inhalation) of a drug influences bioavailability, the fraction of the active form of a drug that enters the bloodstream and successfully reaches its target site. When a drug is given intravenously, absorption is not required, and bioavailability is 100
Conclusion
Pharmacokinetics, sometimes described as what the body does to a drug, refers to the movement of drug into, through and out of the body the time course of its absorption, bioavailability, distribution, metabolism, and excretion. Liberation means the process of release of a drug from the pharmaceutical formulation. Pharmacokinetics of a drug depends on patient-related factors as well as on the drug’s chemical properties. Absorption affects the speed and concentration at which a drug may arrive at its desired location of effect, e.g., plasma. The process of absorption also often includes liberation, or the process by which the drug is released from its pharmaceutical dosage form. Metabolism is the conversion of generally more lipophilic xenobiotic compounds to hydrophilic metabolites that can be eliminated from the body via excretion. The pharmacokinetic measure used to indicate the pattern of distribution of a drug in plasma and in the different tissues, as well as the size of the compartment into which a drug would seem to have distributed in relation to its concentration in plasma, is known as the apparent volume of distribution.
Abbreviations
ATP: Adenosine triphosphate; Cyp450: Cytochrome P450; L/ADME: Liberation/absorption, distribution, biotransformation/metabolism and excretion; CL: Clearance; Ke: elimination rate constant; PDC: Plasma drug concentration; Pgp: P-glycoprotein; PK: Pharmacokinetics; T1/2: Half-life; Vd: Volume of distribution;
Acknowledgments
The author would be grateful to anonymous reviewers by the comments that increase the quality of this manuscript.
Data Sources
Sources searched include Google Scholar, Research Gate, PubMed, NCBI, NDSS, PMID, PMCID, Scopus database, Scielo and Cochrane database. Search terms included: about pharmacokinetics
Funding
None
Availability of data and materials
The datasets generated during the current study are available with correspondent author.
Competing interests
The author has no financial or proprietary interest in any of material discussed in this article.
References
- Ahammad F, Alam R, Mahmud R, Akhter S, Talukder EK, Tonmoy AM, Fahim S, Al-Ghamdi K, Samad A, Qadri I. (2021) Pharmacoinformatics and molecular dynamics simulation-based phytochemical screening of neem plant (Azadiractha indica) against human cancer by targeting MCM7 protein. Brief. Bioinform.
View at Publisher | View at Google Scholar - Feghali M, Venkataramanan R, Caritis S. (2015) Pharmacokinetics of drugs in pregnancy. InSeminars in perinatology (Vol. 39, No. 7, pp. 512-519). WB Saunders.
View at Publisher | View at Google Scholar - Vrbanac J, Slauter R. (2017) ADME in drug discovery. InA Comprehensive Guide to Toxicology in Nonclinical Drug Development (pp. 39-67). Academic Press.
View at Publisher | View at Google Scholar - Shakil Ahmed Saghir, Rais Ahmad Ansari, (2018) in Reference Module in Biomedical Sciences,
View at Publisher | View at Google Scholar - Bisswanger H. (2017) Enzyme kinetics: principles and methods. John Wiley & Sons;
View at Publisher | View at Google Scholar - Shanbhag TV, Shenoy S, Nayak V. (2021). Pharmacology for Dentistry E-Book. Elsevier Health Sciences;
View at Publisher | View at Google Scholar - Holford N, Ma G, Metz D. (2020) TDM is dead. Long live TCI!. British Journal of Clinical Pharmacology.
View at Publisher | View at Google Scholar - Derek G. Waller Anthony P. Sampson , (2018) in Medical Pharmacology and Therapeutics (Fifth Edition),
View at Publisher | View at Google Scholar - Soldin OP, Mattison DR. (2009) Sex differences in pharmacokinetics and pharmacodynamics. Clinical pharmacokinetics. 48(3):143-157.
View at Publisher | View at Google Scholar - Loftsson, T. (2015) “Physicochemical Properties and Pharmacokinetics.” Essential Pharmacokinetics, Pages 85-104.
View at Publisher | View at Google Scholar - Parkinson et al. (2018) “Biotransformation of Xenobiotics”Casarett & Doull’s Toxicology, The Basic Science of Poisons Ninth Edition. Mcgraw-Hill Education Page 194.
View at Publisher | View at Google Scholar - McKenna MJ. (2020) The role of studies of absorption, metabolism, distribution and elimination in animal selection and extrapolation. InHuman Risk Assessment—The Role of Animal Selection and Extrapolation (pp. 113-128). CRC Press.
View at Publisher | View at Google Scholar - Slørdal L,Spigset O, (2005) [Basic pharmacokinetics--absorption]. Tidsskrift for den Norske laegeforening : tidsskrift for praktisk medicin, ny raekke.
View at Publisher | View at Google Scholar - Kalyane D, Raval N, Maheshwari R, Tambe V, Kalia K, Tekade RK. (2019) Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Materials Science and Engineering: C. 1;98:1252-1276.
View at Publisher | View at Google Scholar - Abuhelwa AY, Williams DB, Upton RN, (2017) Foster DJ. Food, gastrointestinal pH, and models of oral drug absorption. European journal of pharmaceutics and biopharmaceutics. 1;112:234-248.
View at Publisher | View at Google Scholar - Yadav A, Mohite S. (2020) Recent advances in protein and peptide drug delivery. Res. J. Pharma. Dosage Forms and Tech. 2020; 12 (3): 205. 1;212.
View at Publisher | View at Google Scholar - Rossi Sebastiano M, Doak BC, Backlund M, Poongavanam V, Over B, Ermondi G, Caron G, Matsson P, Kihlberg J. Impact of dynamically exposed polarity on permeability and solubility of chameleonic drugs beyond the rule of 5. Journal of medicinal chemistry. 2018 Apr 2;61(9):4189-4202.
View at Publisher | View at Google Scholar - Luan X, Zhang LJ, Li XQ, Rahman K, Zhang H, Chen HZ, Zhang WD. (2020) Compound-based Chinese medicine formula: From discovery to compatibility mechanism. Journal of ethnopharmacology. 23;254:112687.
View at Publisher | View at Google Scholar - Min KA, Rosania GR. (2021) Measurement of Transcellular Transport Rates and Intracellular Drug Sequestration in the Presence of an Extracellular Concentration Gradient. InQuantitative Analysis of Cellular Drug Transport, Disposition, and Delivery (pp. 3-39). Humana, New York, NY.
View at Publisher | View at Google Scholar - Behl T, Kaur I, Sehgal A, Kumar A, Uddin MS, Bungau S. (2021) The Interplay of ABC Transporters in Aβ Translocation and Cholesterol Metabolism: Implicating Their Roles in Alzheimer’s Disease. Molecular Neurobiology. 58(4):1564-1582.
View at Publisher | View at Google Scholar - Vasiliou V, Vasiliou K, Nebert DW. (2009) Human ATP-binding cassette (ABC) transporter family. Human genomics. 3(3):1-0.
View at Publisher | View at Google Scholar - Saunders NR, Habgood MD, Møllgård K, Dziegielewska KM. (2016) The biological significance of brain barrier mechanisms: help or hindrance in drug delivery to the central nervous system?. F1000Research. 5.
View at Publisher | View at Google Scholar - Shan N, Perry ML, Weyna DR, Zaworotko MJ. (2014) Impact of pharmaceutical cocrystals: the effects on drug pharmacokinetics. Expert opinion on drug metabolism & toxicology. 10(9):1255-1271.
View at Publisher | View at Google Scholar - Batchelor HK, Marriott JF. (2015) Formulations for children: problems and solutions. British journal of clinical pharmacology. 79(3):405-418.
View at Publisher | View at Google Scholar - Williams AC. (2018) Topical and transdermal drug delivery. Aulton's Pharmaceutics: The Design and Manufacture of Medicines. 5th ed. Edinburgh: Elsevier. 715-725.
View at Publisher | View at Google Scholar - Johnson AR, Forster SP, White D, Terife G, Lowinger M, Teller RS, (2021) Barrett SE. Drug eluting implants in pharmaceutical development and clinical practice. Expert Opinion on Drug Delivery. 10:1-7.
View at Publisher | View at Google Scholar - Pavlović N, Goločorbin-Kon S, Ðanić M, Stanimirov B, Al-Salami H, Stankov K, Mikov M. Bile (2018) acids and their derivatives as potential modifiers of drug release and pharmacokinetic profiles. Frontiers in pharmacology. 8;9:1283.
View at Publisher | View at Google Scholar
