

Research | DOI: https://doi.org/10.31579/2835-835X/079
Molecular Structure Analysis of Morpholin-4-ium p-Aminobenzoate (MPABA) with Antiallergic Effects
1 Kocaeli University, Ali Rıza Veziroğlu Vocational School, Marketing Program, 41780 Kocaeli, Turkey.
2 Istanbul Medeniyet University, Department of Civil Engineering, Uskudar, 34700, Istanbul, Turkey.
*Corresponding Author: Hacer Gümüş, Kocaeli University, Ali Rıza Veziroğlu Vocational School, Marketing Program, 41780 Kocaeli, Turkey
Citation: Hacer Gümüş and Cengiz Ipekb, (2024), Molecular Structure Analysis of Morpholin-4-ium p-Aminobenzoate (MPABA) with Antiallergic Effects, Clinical Trials and Case Studies, 3(5); DOI:10.31579/2835-835X/079
Copyright: © 2024, Hacer Gümüş. This is an open-access artic le distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 17 August 2024 | Accepted: 26 August 2024 | Published: 03 September 2024
Keywords: Molecular docking; NLO; DFT and MEP
Abstract
In order to better understand the molecular definition of the morpholine-4-ium p-aminobenzoate (MPABA) molecule, the physicochemical mechanisms underlying the protein-ligand interaction should be examined in detail. Firstly, Gaussian 09 program was used to determine the ground state molecular geometry of MPABA molecule. Later, Calculations of all molecular and atomic properties such as theoretical vibration spectra (IR), electrical and electronic analysis, electrostatic potential maps and charges were performed by DFT method. Finally, we were investigated by molecular docking method of morpholin with the PDB 6LU7 main protease.
1.Introduction
P-aminobenzoic acid (PABA) [1], also known as vitamin B10, is an organic compound found in some foods and produced by the chemical industry (azo dyes, salts, folic acid, and target esters). The Morpholin-4-ium p-aminobenzoate (MPABA) molecule is a new crystal from the PABA (p-aminobenzoic acid) family. The molecular structure of the MPABA molecule, a synthetic organic compound, contains an azo group (-N=N-). The MPABA molecule forms the basis for the production of azo dyes used in the textile industry. Additionally, the MPABA molecule acts as a bacterial cofactor in folic acid [2] synthesis. The entrance of sulfonamide to the active site on the enzyme [3] surface instead of PABA disrupts the activity of the enzyme and prevents the synthesis of dihydrofolic acid [4]. In order for competitive inhibition to occur (preventing the conversion of PABA to folic acid), a large number of sulfonamide molecules are required [5]. Furthermore, MPABA has shown potential antiallergic effects, making it a compound of interest in the study of allergic reactions.
The crystal structure of morpholin-4-ium p-aminobenzoate (MPABA) [C4H10NO+, C7H6NO2-] was studied by G.Shanmugamet al. in 2012 [1]. G.Shanmugam et al. synthesized [1] the MPABA molecule and its molecular structural, thermal, vibrational and UV–visible transmission analyses were investigated experimentally. However, theoretical calculations of the experimental data for the MPABA molecule were not performed. In this article, we calculated the theoretical data of the MPABA molecule in the Gaussian 09 program to eliminate this deficiency. In addition; Molecular docking calculations were performed with PDB 6LU7 main protease.
2. Computational Details
2.1. DFT calculations
The molecular simulation modeling of the MPABA molecule were calculated using DFT method in Gaussian 09W package [6]. Output data files were visualized using the Gaussian View 5 program [7]. The molecular geometric parameters, IR spectra analysis, molecular frontier orbital energies, molecule charges, molecular docking, electrostatic potential (MEP) surface, electronic and electric properties for the MPABA molecule were calculated using B3LYP and HSEH1PBE levels with 6-311++G(d,p) basis set of DFT method [8].
2.2. Molecular Docking Calculations (Ligand and target protein preparation)
Before starting the molecular docking calculations, the 3D molecular structure of PDB 6LU7 were downloaded from the Protein Data Bank (PDB) of the Structural Bioinformatics Research Laboratory (RCSB) [9]. Thus, the ligand (MPABA) and target (PDB 6LU7) were determined for molecular docking calculations. Molecular Docking calculations were performed by using the AutoDock Tools (ADT) Version 1.5.6 [10] to find the ligand-protein docking interactions. The PyMOL software package [11] has analyzed the output of the AutoDock (version 4.0) program [12]. In addition, Discovery Studio Visualizer 3.5 software [13] was used to visualize the docked active sites in the protein and its H-bond interactions.
3. Results and Discussion
3.1. Geometric optimization
The MPABA molecule was studied by G.Shanmugamet et al. [1]. The unit cell parameters a=5.948(5), b=18.033(4), c=10.577(5) (in Å) β=90.40(1)°and V=1134.5(11) Å3. The numbering of atoms is shown in Figure. 1a [1] and the obtained geometric structure is seen in Fig. 1b.
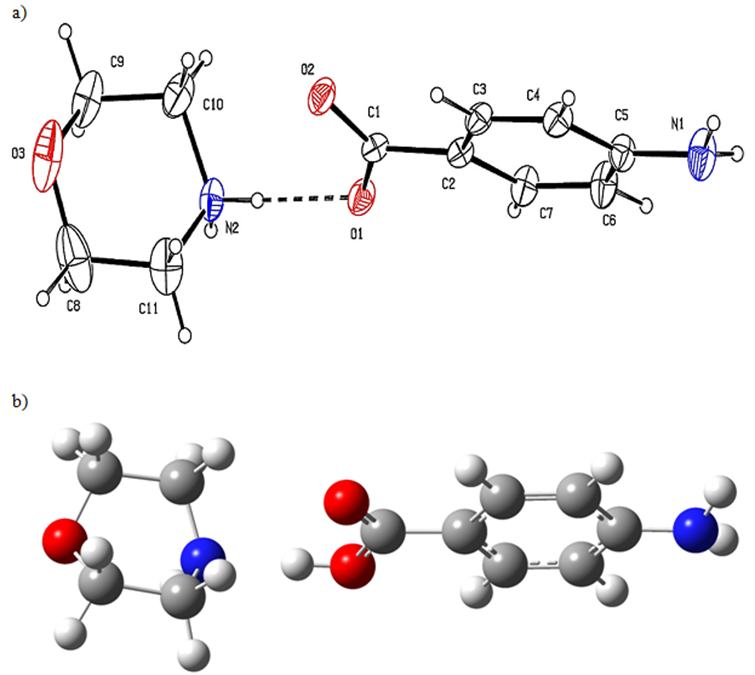
Figure 1 a: The experimental structure b the optimized structure of the MPABA molecule.
Obtained geometric data listed in Table 1. In the study, Theoretical bond lengths (for C-C) were found in the range 1.38140-1.53829 and 1.38493-1.55749 (in Å) at HSEh1PBE and B3LYP, respectively. Experimental bond lengths (C-C) are seen in the range of 1.368(5) and 1.542(9) (in Å) [1]. Experimental C1-O2 and C10-N2 bond lengths are 1.247(4) and 1.493(6) Å, respectively [1]. Calculated bond lengths were observed as 1.22154, 1.47470 Å for the B3LYP and 1.21915, 1.47335 Å for the HSEh1PBE. The experimental O2-C1-O1 bond angle is 123.2(3) ˚[1], and this angle has been seen at 122.81331˚ for the B3LYP and 123.01904˚ for the HSEh1PBE. A good agreement was observed between the theoretical and experimental results for the MPABA molecule, as seen in Table 1. The reason for the differences is that the experimental data is made in the solid phase and the theoretical data is in the gas phase.
| Parameters | Experimental | Theoretical Calculations | |
| X-Ray | DFT/B3LYP | DFT/ HSEH1PBE | |
| Bond lenght(Å) | |||
| C1-O2 | 1.247(4) | 1.22154 | 1.21915 |
| C1-O1 | 1.270(4) | 1.33788 | 1.32677 |
| C1-C2 | 1.498(5) | 1.48356 | 1.47810 |
| C2-C7 | 1.384(5) | 1.40105 | 1.39657 |
| C2-C3 | 1.395(5) | 1.40082 | 1.39627 |
| C3-C4 | 1.373(5) | 1.38493 | 1.38140 |
| C4-C5 | 1.391(5) | 1.40610 | 1.40273 |
| C5-N1 | 1.380(5) | 1.38854 | 1.38075 |
| C5-C6 | 1.389(5) | 1.40530 | 1.40191 |
| C6-C7 | 1.368(5) | 1.38652 | 1.38295 |
| C8-O3 | 1.424(11) | 1.42358 | 1.41706 |
| C8-C11 | 1.542(9) | 1.55748 | 1.53829 |
| C9-O3 | 1.385(8) | 1.42358 | 1.41268 |
| C9-C10 | 1.511(7) | 1.55749 | 1.53585 |
| C10-N2 | 1.493(6) | 1.47470 | 1.47335 |
| C11-N2 | 1.463(7) | 1.47478 | 1.46370 |
| Bond Angles (o) | |||
| O2-C1-O1 | 123.2(3) | 122.81331 | 123.01904 |
| O2-C1-C2 | 120.1(3) | 123.41257 | 123.11416 |
| O1-C1-C2 | 116.7(3) | 113.77412 | 113.86680 |
| C7-C2-C3 | 117.3(3) | 118.54746 | 118.68192 |
| C7-C2-C1 | 120.9(3) | 122.41079 | 122.31850 |
| C3-C2-C1 | 121.8(3) | 119.04175 | 118.99958 |
| C4-C3-C2 | 121.2(3) | 120.99595 | 120.94145 |
| C3-C4-C5 | 121.0(3) | 120.45139 | 120.42385 |
| N1-C5-C6 | 120.5(4) | 120.69049 | 120.67556 |
| N1-C5-C4 | 121.6(4) | 120.64005 | 120.62740 |
| C6-C5-C4 | 117.9(3) | 118.62006 | 118.64929 |
| C7-C6-C5 | 120.8(4) | 120.55201 | 120.52300 |
| C6-C7-C2 | 121.9(3) | 120.83302 | 120.78025 |
| O3-C8-C11 | 111.0(6) | 110.89660 | 111.26917 |
| O3-C9-C10 | 110.5(5) | 110.90485 | 109.90714 |
| N2-C10-C9 | 109.7(5) | 111.68970 | 111.07601 |
| N2-C11-C8 | 108.3(4) | 111.68970 | 111.06367 |
| C11-N2-C10 | 113.7(4) | 110.90437 | 110.24816 |
| C9-O3-C8 | 110.0(4) | 110.98402 | 110.82419 |
| Dihedral Angles(o) | |||
| O2-C1-C2-C7 | -163.0(4) | -179.95412 | 179.72080 |
| O1-C1-C2-C7 | 16.1(5) | 0.05007 | -0.26649 |
| O2-C1-C2-C3 | 13.6(5) | 0.02179 | -0.30592 |
| O1-C1-C2-C3 | -167.3(3) | -179.97401 | 179.70679 |
| C7-C2-C3-C4 | -0.8(5) | 0.02004 | 0.05006 |
| C3-C4-C5-C6 | 0.0(6) | -0.12400 | -0.15276 |
| N1-C5-C6-C7 | 178.3(4) | 177.57408 | 177.68626 |
| C4-C5-C6-C7 | -1.8(7) | 0.11422 | 0.18369 |
| C5-C6-C7-C2 | 2.4(7) | 0.05794 | -0.09930 |
| C3-C2-C7-C6 | -1.0(6) | -0.02999 | -0.01900 |
| C1-C2-C7-C6 | 175.7(4) | 179.94604 | 179.95437 |
| O3-C9-C10-N2 | 56.7(6) | -2.98118 | -29.36309 |
| O3-C8-C11-N2 | -55.4(7) | 3.20928 | -23.80272 |
| C8-C11-N2-C10 | 49.8(7) | 51.01198 | 61.53772 |
| C9-C10-N2-C11 | -51.0(6) | -51.13477 | -33.49511 |
| C10-C9-O3-C8 | -64.3(7) | 59.60338 | 69.69971 |
| C11-C8-O3-C9 | 64.0(6) | -59.72907 | -40.79314 |
Table 1 Geometric parameters for the MPABA molecule.
3.2. IR Spectra Analysis
G.Shanmugamet et al. [1] experimentally reported spectrum and band vibrations in the 4000-400 cm-1 Mid-infrared (IR) region for the MPABA molecule [1]. In this study, theoretical vibration frequencies were calculated for MPABA molecule at DFT method. The values calculated in the gas phase at the B3LYP/6311++G(d,p) level were multiplied by a coefficient of 0.9970 in order to be compatible with the experimental values in the solid phase. In the HSEH1PBE/6311++G(d,p) method, it was multiplied with a coefficient of 0.9614 [8]. The harmonic vibration frequencies and assignments of the MPABA molecule multiplied by the scale factors are listed in Table 2.
| Assigments | Exp. (cm-1) | B3LYP | HSEH1PBE | ||
| N–H stretching (asymmetric) in NH2+ cation | 3442.68 | 3670.70 | 3581.46 | ||
| N–H stretching (symmetric) in NH2+ cation | 3312.99 | 3568.95 | 3477.85 | ||
| Morpholinium ring CH2 stretch (asymmetric) | 2997.24 | 3075.53 | 2974.77 | ||
| Morpholinium ring CH2 stretch (symmetric) | 2765.97 | 2968.15 | 2881.60 | ||
| Stretching of C=O | 1700.01 | 1725.73 | 1695.72 | ||
| Aromatic ring C=C stretch | 1715.24 | 1658.30 | 1617.04 | ||
| Asymmetric carboxylate anion (COO-) stretch | 1597.01 | 1604.66 | 1573.80 | ||
| Symmetric carboxylate anion (COO-) stretch | 1366.17 | 1356.55 | 1338.89 | ||
| C–H inplane bending/Asymmetric C–O–C stretch | 1167.39 | 1142.13 | 1114.14 | ||
| Symmetric C–O–C stretch | 1090.81 | 1114.42 | 1094.78 | ||
| C–N inplane bending | 999.70 | 1010.29 | 982.782 | ||
| Aromatic C–H out of plane bending | 835.89 | 848.168 | 825.343 | ||
| C–N–H bending | 775.66 | 784.210 | 764.525 | ||
| CH2 rocking | 688.28 | 730.352 | 662.703 | ||
| N–H out of plane bending | 610.44 | 622.995 | 605.490 | ||
| C–N out of plane bending | 527.29 | 546.785 | 522.031 | ||
| Out of plane ring C=C bending | 442.30 | 422.180 | 407.095 | ||
Table 2 Harmonic vibrational wavenumbers and assignments for the MPABA molecule.
As seen in Table 2, the C-H2 asymmetric stretching vibration band was experimentally observed at 2997 cm-1, while the theoretical vibration frequencies were calculated as 3076 cm-1 in the B3LYP method and 2975 cm-1 in the HSEH1PBE method. While the N-H symmetrical stretching vibration band was observed experimentally at 3313 cm-1, it was theoretically calculated as 3569 cm-1 in the B3LYP method and 3478 cm-1 in the HSEH1PBE method. While C=O stretching vibration bands were observed experimentally at 1700 cm-1, they were calculated as 1726 cm-1 in the B3LYP method and 1696 cm-1 in the HSEH1PBE method. It is seen that the computational vibration frequencies with the local and gradient corrected DFT method are compatible with the experimental frequencies. In addition, the values calculated by the HSEH1PBE method are closer to the experimental values. The experimental [1] and calculated harmonic vibration frequencies for the MPABA molecule are shown in Figure 2.
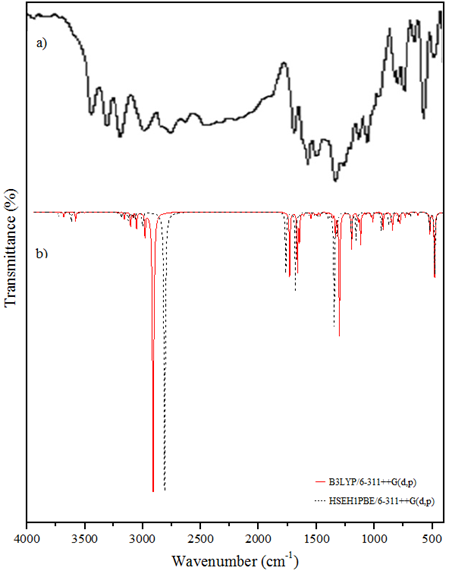
Figure 2: a FT-IR spectrum b the calculated IR spectra of the MPABA molecule.
3.3. Electronic Properties
In electronics studies, the molecular orbital helps describe the wave-like behavior of electrons within the molecule. It serves to calculate chemical and physical properties such as the probability of finding an electron in any part of the molecule. Molecular orbitals are called HOMO-LUMO. HOMO is the molecule's tendency to donate electrons and is the highest energy orbital that is occupied. LUMO, on the other hand, is the molecule's tendency to gain electrons and is the lowest vacant orbital. HOMO and LUMO are called Frontier Molecular Orbitals because they serve as parent molecular orbitals in chemical reactions. AMolecular orbitals can be calculated quantitatively using quantum chemical calculation methods (such as HF, DFT). EHOMO and ELUMO energy values of MPABA molecule were calculated using the DFT method. By looking at the electron density distribution of the calculated EHOMO and ELUMO energies, the energy difference (ΔE), ionization potential (I), electron affinity (A), electronegativity (χ), chemical hardness (η) and chemical softness (S) parameters are provided by formulas and the results are given in Table 3.
| B3LYP | HSEH1PBE | |
| EHOMO (eV) | -5.94 | -5.72 |
| ELUMO (eV) | -0.95 | -1.07 |
| ΔE = ELUMO-EHOMO (eV) | 4.99 | 4.65 |
| I (eV) | 5.94 | 5.72 |
| A (eV) | 0.95 | 1.07 |
| c (eV) | 3.45 | 3.40 |
| h (eV) | 2.50 | 2.33 |
| S (eV-1) | 0.20 | 0.22 |
| ETOTAL (a.u) | -764.21787101 | -763.40197626 |
Table 3: The electronic properties for the MPABA molecule.
In the B3LYP method, the HOMO and LUMO energies for the MPABA molecule are -5.94 and -0.95 eV, and the energy gap between the HOMO and LUMO orbitals is 4.99 eV. It is well known that lower value for energy difference (ΔE) of MPABA molecule makes it more polarized, more reactive and less stable. Also called soft molecule. In the 3D plots shown in Figure 3, positive charge red color and negative charge green color are represented.
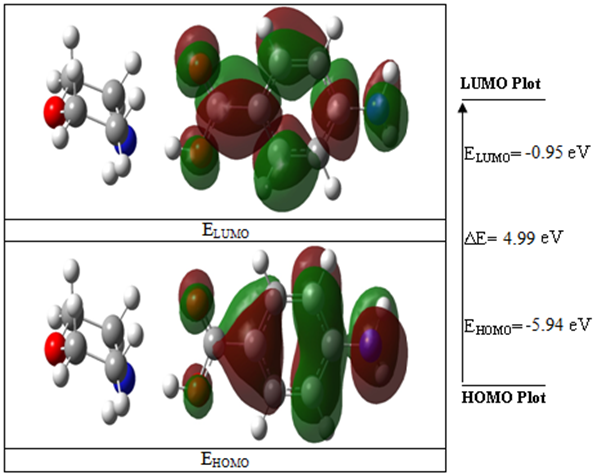
Figure 3: Molecular frontier orbital energy pictures of the MPABA molecule.
3.4. Electric Properties
Organic and inorganic molecules exhibit nonlinear optical (NLO) properties due to polarizability. Polarity (<α>) is defined as the response of the static dipole moment of the molecule to an applied external electric field. Polarizability, which is a measure of the molecule's properties such as electron distribution and charge density, is an important quantity used in molecular spectroscopy. If it causes deformation of the molecule independent of the direction of the applied electric field, it is called isotropic polarizability. If it depends on the direction of the applied electric field, it is called anisotropic (Δα) polarizability. The dipole moment (µ) is an important quantity in molecular chemistry as it describes the intramolecular charge movement. Nonlinear optical properties were calculated for the MPABA molecule using the B3LYP and HSEH1PBE methods and are listed in Table 4.
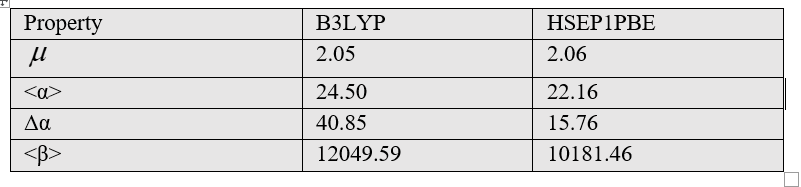
Table 4 The electric properties for the MPABA molecule.
As seen in Table 4, the dipole moment was calculated as 2.05 Debye in the B3LYP method and 2.06 Debye in the HSEH1PBE method with the help of formulas [10]. The anisotropy of the polarizability of the MPABA molecule was calculated as 15.76×10-24esu in HSEH1PBE method with the help of formulas [14]. The polarizability result of MPABA molecule was calculated 22.16×10-24esu in HSEH1PBE method with the help of formulas [14]. The first-order hyperpolarizability result of MPABA molecule was calculated 10181.46×10-33esu in HSEH1PBE method with the help of formulas [14].
3.5 Molecular electrostatic potential (MEP) maps and Atomic charge analysis
MEP provides information about the local polarity of the molecule. MEP surface can be calculated especially for large molecules; it is also used to express steric interactions between polarized regions of molecular shape and often biomolecules [15]. MEP surface map is very important for the investigation of molecular structure. MEP surface map shows the molecule's color, shape, charge, size, and electrostatic potential regions at the same time. The color chart of MEP is red for (-) charge, blue for (+) charge, yellow for the slightly electron-rich region, green for neutral [16]. The MEP maps of the MPABA molecule was calculated at B3LYP method and the polarization effect is clearly visible in Figure 4.
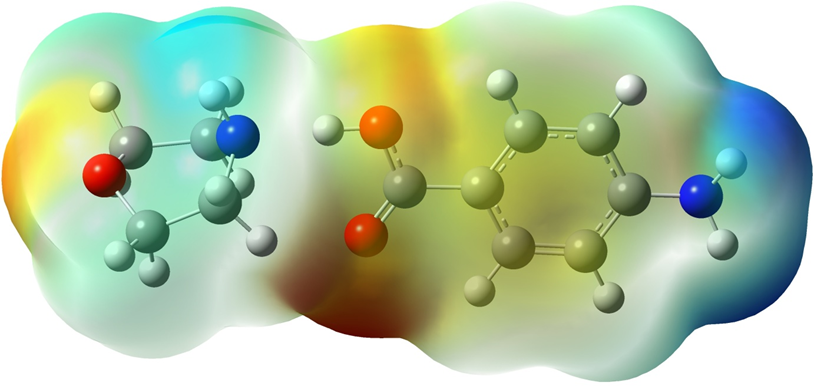
Figure 4: Molecular electrostatic potential surface maps of MPABA molecule.
Mulliken charge distribution is an old method and common method [34]. In case making the distribution to atoms of wave functions, it is based on the principle of equally distributing two overlapping orbits. In order to better understand the interatomic interactions of the MPABA molecule, the atomic charges of the molecules were calculated by the B3LYP and HSEH1PBE methods and listed in Table 5.
| Atom | B3LYP | HSEH1PBE |
| C8 | -0.14001 | -0.192 |
| C11 | 0.27917 | 0.25702 |
| C10 | 0.2826 | 0.25651 |
| C9 | -0.14311 | -0.19238 |
| O3 | -0.15183 | -0.09435 |
| N2 | -0.17016 | -0.08012 |
| C3 | -0.06122 | -0.07501 |
| C4 | -0.21324 | -0.22869 |
| C5 | 0.08599 | 0.04835 |
| C6 | -0.35087 | -0.37578 |
| C7 | -0.19188 | -0.20018 |
| C2 | 0.50776 | 0.561 |
| N1 | 0.16328 | 0.18871 |
| C1 | 0.14663 | 0.12907 |
| O2 | -0.25319 | -0.24607 |
| O1 | 0.21007 | 0.2439 |
Table 5: The Mulliken charge distribution calculated for the MPABA molecule.
The magnitude of C charges is given to be either (+) or (-) are recorded to change from -0.35087 to 0.50776 with the B3LYP method in Table 5. Mulliken charge on each atom of the MPABA molecule is shown in Figure 5.
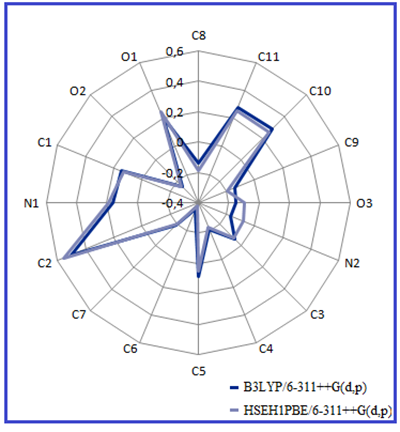
Figure 5: The Mulliken charges of the MPABA molecule.
3.8. Molecular Docking
Proteins are biological macromolecules that perform an important part of vital functions, as well as structurally. Proteins perform their functions by binding to proteins or other molecules like themselves. Protein-ligand interactions must be studied to understand biology at the molecular level. Knowledge of the protein-ligand interaction system and the mechanisms responsible for their binding plays an important role in the discovery, design, and development of drugs. Molecular docking was performed by AutoDock (version 4.0) program. The program receives a semi-empirical free energy force in docking simulation processes. The force field contains (Vi) and a loss of conformation alenthropy after binding (ΔSconf):
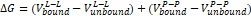
where L refers to the ligand and Prefers to the protease [17]. 3D molecular interaction diagrams of target protease (PDB 6LU7) and ligand (MPABA) are given in Figure 6.
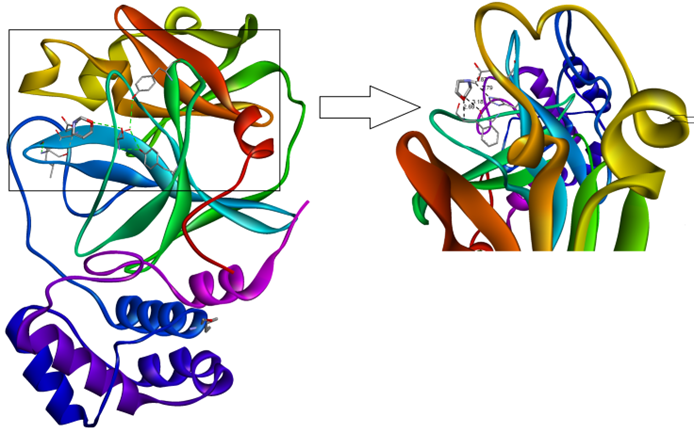
Figure 6: The 3D interaction diagrams of the MPABA molecule in into the active sites PDB 6LU7 main protease.
Detailed 2D interaction diagrams are seen in Figure 7. The MPABA molecular docking is examined in activesites of the PDB 6LU7 main protease. The PDB 6LU7 target protease exhibits the minimum binding energy of -2.6 kcal/mol, intermolecular energy of -3.43 kcal/mol, and an inhibition constant of 12.37 micromolar (mM). The deviation between the ligand- protease is analyzed, where the root mean square deviation (RMSD) value is calculated as 15.36 for MPABA. Molecular docking results show that the synthesized Morpholin complex has conventional hydrogen bond interactions with residue A140-A166-A172, which is characterized by an interaction length of 1.87-2.79 A of PDB 6LU7, and at the same time, carbon hydrogen bond interactions with residue A138 with an interaction length of 3.18 A. bond interaction. The total energy of the best conformation obtained for the morpholin complex was calculated as 3.43 kcal/mol, and the inhibition constant was calculated as 12.37 µM (micromolar).According to these results, it is seen that Morpholin synthesis is semi-active with complex PDB 6LU7.
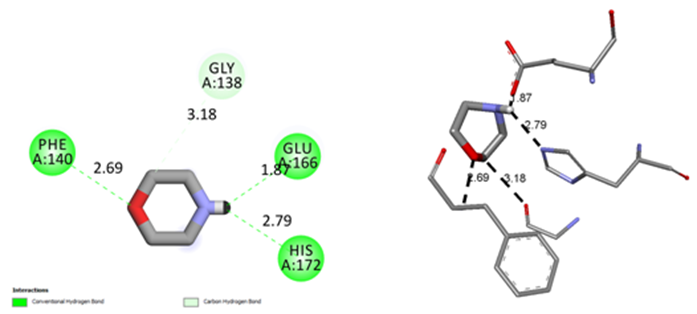
Figure 7: The binding orientation and 2D molecular interaction diagrams of MPABA molecule in into the active sites of PDB 6LU7 main protease.
Conclusion
In order to have more detailed information about the protein-ligand interactions, we examined all the physicochemical properties of the MPABA molecule. Firstly, the geometry of the MPABA molecule was optimized at different levels using the DFT/B3LYP and DFT/HSEH1PBE methods with the 6-311++G(d,p) basis set. The calculated structural parameters, such as bond distances, bond angles, and dihedral angles, compared well with the experimental values. Additionally, the calculated IR spectra and the assignments made were compared with the experimental values.
Moreover, the frontier molecular orbital energies and the energy gap between HOMO and LUMO were calculated. The observed small energy gap indicated that charge transfer occurs in MPABA. The compound exhibits strong effective intra- and intermolecular charge transfer and shows a mean first hyperpolarizability (β = 10181.46 × 10⁻³³ esu). The MEP plot clearly demonstrated the reactive sides, as well as the electrophilic and nucleophilic attack regions.
Finally, the molecular interaction of MPABA with the target proteins was docked, and the minimum binding energy value was examined. The active site binding properties and binding conformations were visually observed through the molecular docking study. Morpholin-4-ium p-aminobenzoate (MPABA) is a synthetic organic compound with an (-N=N-) azo group in its molecular structure and is produced through synthesis. The MPABA molecule forms the basis for the production of azo dyes used in the textile industry. Furthermore, due to its potential antiallergic effects, it aims to contribute to the textile industry by being incorporated into fabrics designed to minimize allergic reactions.
References
- Shanmugam, G., Kumar, K. R., Sridhar, B., and Brahadeeswaran, S., (2012). Synthesis, structure, growth and characterization of a novel organic NLO single crystal: Morpholin-4-ium p-aminobenzoate. Mater. Res. Bull. 47: 2315-2323.
View at Publisher | View at Google Scholar - Robinson, F. A. (1966). The vitamin Co-factors of Enzyme Systems. Pergamon, London, 541: 1-499.
View at Publisher | View at Google Scholar - Pauling, L., Hayward, R., and Freeman, W. H. (1964). The Architecture of Molecules. San Francisco, 56.
View at Publisher | View at Google Scholar - Gracin, S., and Rasmuson, A. C. (2004). Polymorphism and Crystallization of p-Aminobenzoic Acid. Cryst. Growth Des. 4, 1013-1023.
View at Publisher | View at Google Scholar - Etter, M.C., and Frankenbach, G.A. (1989). Hydrogen-bond directed cocrystallization as a tool for designing acentric organic solids. Chem. Mater. 1,: 10-12.
View at Publisher | View at Google Scholar - Frisch M.J. et al. 2009. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT.
View at Publisher | View at Google Scholar - GaussView, Version 5, R Dennington, T Keith and J Millam (Shawnee Mission, KS: Semichem Inc) 2009.
View at Publisher | View at Google Scholar - Altürk, S., Avcı D., Tamer Ö., Atalay Y., 1Hepyrazolee3ecarboxylic acid: Experimental and computational study, Journal of Molecular Structure 1164 (2018) 28-36.
View at Publisher | View at Google Scholar - http://www.rcsb.org/
View at Publisher | View at Google Scholar - Sanner, M. F. Python: A Programming Language for Software Integration and Development. Journal of Molecular Graphics and Modelling 1999, 17, 57-61.
View at Publisher | View at Google Scholar - De Lano, W. L. (2004) The PyMOL Molecular Graphics System; De Lano Scientific: San Carlos, CA, 2004.
View at Publisher | View at Google Scholar - Morris, G. M.; Huey, R.; Lindstrom, W.; Sanner, M. F.; Belew, R. K.; et.al (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry 2009, 30, 2785-2791.
View at Publisher | View at Google Scholar - Dassault Syst_emes BIOVIA DiscoveryStudio Modeling Environment. Release 2017 Dassault Systemes, 2016.
View at Publisher | View at Google Scholar - Eşme A.: Spectroscopic calculations, Hirshfeld surface analysis, and molecular docking studies of anticancer 6-(4-Aminophenyl)-4-(4-methoxyphenyl)-2-methoxynicotinonitrile. Spect. Lett. 54, 51-64 (2021).
View at Publisher | View at Google Scholar - Gümüş, H.P.: Conformational, spectroscopic, electric and electronic investigations on 5-nitropyridine-2-hydrazino-3-carbonitrile-6-methyl-4-(methoxymethyl) (molecule 2): Molecular docking study. J. Molecular Struct. 1211, 128018 (2020).
View at Publisher | View at Google Scholar - Khattab, M. and Al-Karmalawy, A. A., Frontiers in Chemistry, Revisiting Activity of Some Nocodazole Analogues as a Potential Anticancer Drugs Using Molecular Docking and DFT Calculations.
View at Publisher | View at Google Scholar - M. Garrett, D.S.G. Morris, E. Michael, Pique, William “Lindy” Lindstrom, R. Huey, Stefano and W.E.H. Forli, Scott Halliday, Rik Belew and Arthur J. Olson, User Guide AutoDock Version 4.2, Automated Docking of Flexible Ligands to Flexible Receptors. 49 (2010).
View at Publisher | View at Google Scholar
