

Research Article | DOI: https://doi.org/10.31579/2835-835X/107
Investigation of the Electronic Structure Analyses of Antioxidant 2,2'-Dithymoquinone by using DFT Method
Automotive Technology Program, Golcuk Vocational School, Kocaeli University, 41380, Kocaeli, Turkey.
*Corresponding Author: 10.31579/2835-835X/107
Citation: Hacer Gümüş, (2025), Investigation of the Electronic Structure Analyses of Antioxidant 2,2'-Dithymoquinone by using DFT Method, Clinical Trials and Case Studies, 4(3); DOI:10.31579/2835-835X/107
Copyright: © 2025, Hacer Gümüş. This is an open-access artic le distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 15 April 2025 | Accepted: 29 April 2025 | Published: 02 May 2025
Keywords: 2,2'-dithymoquinone; dft (density functional theory); electronic structure
Abstract
In this study, the electronic structure of the antioxidant compound 2,2'-Dithymoquinone was investigated using the Density Functional Theory (DFT) method. The molecular geometry of 2,2'-Dithymoquinone was optimized in the gas phase using the Gaussian program. From the optimized structure, the electronic properties were determined, and theoretical evaluations were made regarding the potential biological activities of the compound based on these findings.
1. Introduction
Natural compounds [1], especially their antioxidant [2] properties, hold significant potential in the treatment of diseases associated with oxidative stress. Antioxidants [3] neutralize free radicals, preventing cellular damage and strengthening the body's defense mechanisms. In this context, thymoquinone and its derivatives are among the important compounds that stand out for their antioxidant properties.
2,2'-Dithymoquinone, a dimeric derivative of thymoquinone, exhibits strong antioxidant [4] effects due to its unique bond structures provided by its molecular framework. Understanding the chemical and biological properties of this compound is crucial for potential therapeutic applications. However, the properties of 2,2'-Dithymoquinone have primarily been investigated through experimental methods, with theoretical analyses remaining limited.
The aim of this study is to theoretically investigate 2,2'-Dithymoquinone by performing electronic analyses using the Density Functional Theory (DFT) method, taking into account the compound's antioxidant activity.
2. Computatıonal Detaıls
The geometric structure of the antioxidant compound 2,2'-Dithymoquinone was initially visualized [5] using the Gaussian View [6] program. Subsequently, theoretical calculations were performed using the Density Functional Theory (DFT) method, with the B3LYP [7, 8] functional and the 6-311G(d,p) basis set. This computational approach aimed to investigate the electronic structure of 2,2'-Dithymoquinone.
3. Results And Discussion
3.1. Analysis of Molecular Geometry Structure
The molecular structure of 2,2'-Dithymoquinone was optimized using the Density Functional Theory (DFT) method [9] with the B3LYP/6-311++G(d,p) functional in the gas phase. The molecule is composed of 20 carbon (C), 24 hydrogen (H), and 4 oxygen (O) atoms. 2,2'-Dithymoquinone exhibits a dimeric structure formed by the bonding of two thymoquinone units. It belongs to the P-1 space group and displays a symmetric configuration, which has a significant impact on its electronic properties.
The optimization process provided insights into the molecule’s overall geometry, yielding a stable structure. Geometric optimization plays a key role in accurately evaluating the compound’s potential biological activity, as the correct three-dimensional structure governs its ability to interact with other molecules. Furthermore, this process directly influences the molecule’s energy levels and stability. The optimized structure of 2,2'-Dithymoquinone is presented in Figure 1.
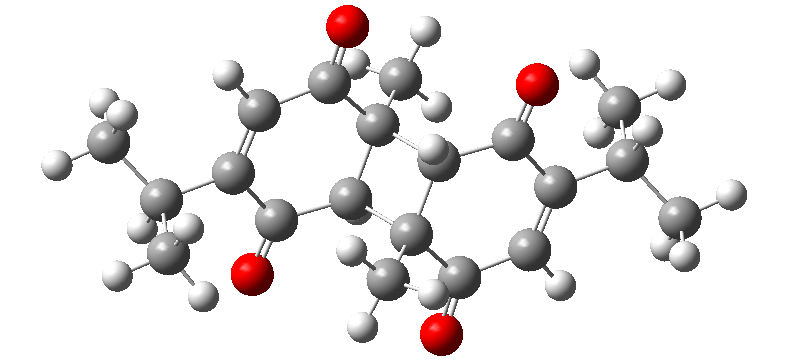
Figure 1. Optimized geometry of 2,2'-Dithymoquinone calculated using the B3LYP/6-311++G(d,p) method
3.2. Analysis of Molecular Electronic Properties
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are fundamental components in understanding a molecule’s reactivity, stability, and interaction behavior. While the HOMO energy reflects the molecule’s electron-donating capability (π-donor), the LUMO energy is related to its electron-accepting ability (π-acceptor). These frontier molecular [10] orbitals provide insights into possible electron transfer processes and are highly relevant for assessing biological activity.
| Table 1. Molecular orbital energy calculations of 2,2'-Dithymoquinone. | |
| Parameters | B3LYP /6-311++G(d,p) |
| EHOMO (eV) | -7.04971 |
| ELUMO (eV) | -3.06838 |
| ΔE = ELUMO-EHOMO (eV) | 3.98133 |
| I (eV) | 7.04971 |
| A (eV) | 3.06838 |
| c (eV) | 5.059045 |
| h (eV) | 1.990665 |
| S (eV-1) | 0.070925 |
| ETOTAL (a.u) | -1077.7311 |
Table : Presents the calculated electronic parameters of 2,2'-Dithymoquinone, which were obtained using Density Functional Theory (DFT) at the B3LYP/6-311++G(d,p) level.
According to the results in Table 1, the HOMO and LUMO energy levels of 2,2'-Dithymoquinone were calculated as –7.04971 eV and –3.06838 eV, respectively. The energy gap (ΔE) of 3.98133 eV indicates moderate reactivity along with good electronic stability, suggesting the compound’s suitability for biological interactions.
The ionization potential (I) and electron affinity (A), determined as 7.04971 eV and 3.06838 eV respectively, reveal the molecule’s ability to participate in redox reactions. Additionally, the chemical potential (–
5.059045 eV), chemical hardness (1.990665 eV), and softness (0.070925 eV⁻¹) further support its potential for selective interaction with electrophilic and nucleophilic species. The total electronic energy of the optimized structure was computed as –1077.7311 atomic units (a.u.), indicating a stable ground-state configuration.
A visual representation of the frontier orbitals is provided in Figure 2, which illustrates the spatial distribution of the HOMO and LUMO regions in the molecule.
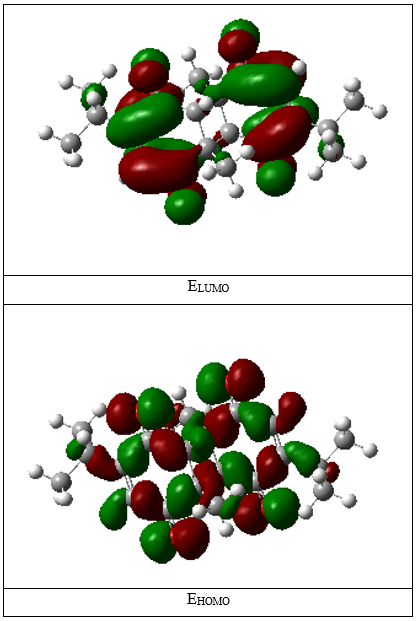
Figure 2. 3D representation of the HOMO and LUMO energy levels of 2,2'-Dithymoquinone.
The spatial distribution of the HOMO and LUMO orbitals provides valuable insight into the reactive sites of the molecule. As visualized in Figure 2, the HOMO orbital is primarily localized over the quinone rings, suggesting that these regions are likely to act as electron-donating centers in redox or charge-transfer processes. In contrast, the LUMO orbital extends across similar π-conjugated regions, indicating the molecule’s propensity to accept electrons in electrophilic interactions.
This orbital alignment supports the hypothesis that 2,2'-Dithymoquinone may act as an effective antioxidant, by stabilizing free radicals through delocalization of unpaired electrons. The moderate HOMO–LUMO energy gap (ΔE = 3.98133 eV) further suggests that the molecule maintains a balance between chemical stability and reactivity an important feature for compounds that participate in redox cycling without undergoing rapid degradation.
Moreover, the values of global reactivity descriptors such as ionization potential (I), electron affinity (A), and chemical softness (S) are consistent with known antioxidant agents. The relatively low chemical hardness (η = 1.990665 eV) and non-zero softness (S = 0.070925 eV⁻¹) imply that the molecule has a flexible electronic structure, which may enhance its interaction with reactive oxygen species or other electrophilic agents in biological systems.
In conclusion, the combination of spatial orbital distribution and energetic properties suggests that 2,2'-Dithymoquinone could play a significant role as an electron donor in biochemical environments, supporting its potential use in therapeutic strategies targeting oxidative stress.
4. Conclusion
In this study, the electronic structure of the antioxidant compound 2,2'-Dithymoquinone was thoroughly investigated using the Density Functional Theory (DFT) method. Geometry optimization performed at the B3LYP/6-311++G(d,p) level revealed that the compound possesses a stable and symmetric structure. Its dimeric nature and molecular symmetry provided important insights into its electronic characteristics.
Analysis of the HOMO and LUMO energy levels enabled predictions about the chemical reactivity and potential biological effects of the molecule. These theoretical findings offer a fundamental basis for evaluating the pharmaceutical and therapeutic potential of 2,2'-Dithymoquinone.
This study complements the predominantly experimental literature on 2,2'-Dithymoquinone by providing theoretical data, and it may serve as a guide for future experimental and computational research on this compound.
References
- Myers, A. L., Zhang, Y.-P., Kramer, M. A., Bornmann, W. G., Kaseb, A., Yang, P., & Tran, H. T. (2012). A practical synthesis and X-ray crystallographic analysis of dithymoquinone, a photodimer of thymoquinone. Letters in Organic Chemistry, 9, 762-766.
View at Publisher | View at Google Scholar - Parcheta, M., Świsłocka, R., Orzechowska, S., Choińska, R., & Lewandowski, W. (2021). Recent developments in effective antioxidants: The structure and antioxidant properties. Materials, 14(7), 1984.
View at Publisher | View at Google Scholar - Magalhães, L. M., Segundo, M. A., Reis, S., & Lima, J. L. F. C. (2008). Methodological aspects about in vitro evaluation of antioxidant properties. Analytica Chimica Acta, 613(1), 1-19.
View at Publisher | View at Google Scholar - Khireddine, A., Boukelkoul, M., Atalay, Y., Tamer, Ö., Avcı, D., Merzoud, L., & Chermette, H. (2022). Structural, electronic, thermodynamic, optical and nonlinear optical properties of curcumin complexes with transition metals: DFT and TD-DFT study. ChemistrySelect, 7, e202104442.
View at Publisher | View at Google Scholar - Frisch M.J. et al. (2009). Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT.
View at Publisher | View at Google Scholar - GaussView, (2009) Version 5, R Dennington, T Keith and J Millam (Shawnee Mission, KS: Semichem Inc).
View at Publisher | View at Google Scholar - Becke, A.D. (1993). J. Chem. Phys. 98, 5648.
View at Publisher | View at Google Scholar - Lee, C., Yang, W., & Parr, R.G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density Phys. Rev. B 37, 785.
View at Publisher | View at Google Scholar - Gümüş, H., Tekin, N., & Kara, Y. S. (2023). Structural and spectroscopic characterization of 3-[4-(trifluoromethyl)phenyl]-3a,4,8,8a-tetrahydro-6H-[1,3] dioxepino[5,6-d][1,2]oxazole compound: An experimental and density functional theory study. Journal of Applied Spectroscopy, 89(6), 1150-1157.
View at Publisher | View at Google Scholar - Gümüş, H.P.: (2020) Conformational, spectroscopic, electric and electronic investigations on 5-nitropyridine-2-hydrazino-3-carbonitrile-6-methyl-4-(methoxymethyl) (molecule 2): Molecular docking study. J. Molecular Struct. 1211, 128018.
View at Publisher | View at Google Scholar
