

Research Article | DOI: https://doi.org/10.31579/2834-5177/046
Investigation of Measles Outbreak in Pakistan (2022 to 2023): Exploring Comorbidities, Complications, and Molecular Dynamics
1Candidate, Medical Library and Information Science, Faculty of Management and Medical Information Sciences, Kerman University of Medical Sciences,Kerman, Iran.
2Associate Professor , Department of Medical Library & Information Sciences, Faculty of Management and Medical Information Sciences, Kerman University of Medical Sciences., Kerman, Iran.
3Associate Professor, Department of Knowledge and Information Science, Shahid Bahonar University of Kerman, Kerman, Iran.
4Assistant professor, Health in Disasters and Emergencies Research Center, Institute for Futures Studies in Health, Kerman University of Medical Sciences, Kerman, Iran.
5Associate Professor, Department of Medical Library & Information Sciences, Faculty of Management and Medical Information Sciences, Kerman University of Medical Sciences., Kerman, Iran.
*Corresponding Author: Muhammad Ali, Candidate, Medical Library and Information Science, Faculty of Management and Medical Information Sciences, Kerman University of Medical Sciences, Kerman, Iran.
Citation: Asia Nawaz, Sidra Rahman, Nighat Haider, Adnan Zeb, Muhammad Ali, (2025), Investigation of Measles Outbreak in Pakistan (2022 to 2023): Exploring Comorbidities, Complications, and Molecular Dynamics, International Journal of Clinical Infectious Diseases, 4(3); DOI:10.31579/2834-5177/046
Copyright: © 2025, Muhammad Ali. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 05 June 2025 | Accepted: 18 June 2025 | Published: 26 June 2025
Keywords: measles; epidemiology; nucleoprotein gene; phylogenetic analysis; b3 genotype; pakistan
Abstract
Abstract
Background
Measles remains a major public health concern worldwide, particularly in regions with insufficient vaccination coverage and healthcare infrastructure like Pakistan. The purpose of this study is to investigate that what are the epidemiological, molecular, and phylogenetic characteristics of the measles outbreak in Pakistan during 2022- 2023 and highlight the areas of improvement that can contribute to the understanding of the disease and inform strategies for effective outbreak management and vaccination optimization.
Methods
Throat swabs were collected from patients (n = 183) admitted at PIMS, Islamabad, Pakistan from December 2022- December 2023. This research was conducted statistically by using SPSS-21 software. The phylogenetics, amino acid substitutions, and structural analysis of partial nucleoprotein were performed by using different bioinformatics software.
Results
Most of the measles patients were infants with a high prevalence of comorbidity and complications and significant patients had inadequate vaccination coverage. The phylogenetic analysis revealed relatedness to B3 strains circulating in Russia and the USA, emphasizing the global spread. The amino acid substitutions and structure analysis highlighted minor structural variations in current isolates as compared to the reference strain.
Conclusion
The findings underscore the importance of continuous surveillance and understanding of measles epidemiology and viral variations for effective outbreak management and vaccination optimization, ultimately aiming to mitigate the morbidity and mortality associated with measles outbreaks in Pakistan and similar settings globally. This study exemplifies the significance of interdisciplinary collaboration in confronting complex infectious disease challenges, emphasizing the imperative of continuous monitoring and preparedness in combating measles
1.Introduction
Measles continues to be a major contributor to the global disease burden. It is a highly infectious disease caused by the Measles virus that belongs to the genus Morbillivirus within the family Paramyxoviridae. The measles virus is serologically monotypic and has been genetically characterized into eight clades (A-H) and 24 genotypes (A, B1-3, C1-2 D1-11, E, F, G1- 3, and H1-2). Molecular and epidemiological studies elucidate the spatiotemporal origin of circulating viruses and highlight any changes in the typical pattern and severity of disease in an endemic region [1]. Despite the availability of an effective measles vaccine for 50 years, it is still causing significant mortality and morbidities among young children in underdeveloped countries specifically targeting the younger population in resource-poor settings [2]. Measles clinically presents itself as fever cough, coryza, maculopapular rash, and conjunctivitis. In severe cases, it can lead to several neurological, gastrointestinal, and respiratory complications and even death [3]. Measles affects all the organs of the body via infecting mucous membranes and immunosuppression due to the prolonged infection renders the patient prone to several secondary infections. The mode of transmission for measles is through respiratory droplets and aerosols and having a high reproduction number (R0) around 12-18 in susceptible populations can be quite contagious [4]. The incubation period of measles is approximately two weeks, and the patient recovers within three weeks if the infection is mild. In other cases when the infection is persistent it can take serious form and lead to severe complications such as pneumonia that is fatal pertinent to 56-86% of measles-related deaths. In around 50% of pneumonia there is bacterial superinfection [5]. Diarrhea and brain infections including encephalitis, and meningitis are also major causes of death among measles patients. Encephalitis is caused by the primary viral infection if the virus makes its way into the brain of an infected person and is followed by chemokines and lymphokines infiltration. In this situation around 10-15% of patients succumb to death and if the virus persists it can lead to post-infection Encephalomyelitis (PIE) that causes permanent damage to the nervous system [6-7].
From 2000 to 2021, around 56 million deaths were averted due to extensive immunization in different measles- endemic regions. In 2021 a mass measles and rubella vaccination campaign was launched in Pakistan that aimed to immunize 90 million children between 9 months to 15 years [8]. As a result, the overall vaccine coverage remained at 81%. During the summer of 2022 Pakistan was hit by major floods that affected the health infrastructure and massive displacements into temporary settlements and camps provided breeding grounds for measle outbreaks that reared its head in 31 districts of Pakistan. According to data from the National Disaster
Management Authority 12 out of 13 (almost 92%) of these measles-endemic areas were flood-affected. By the end of 2022 another measles immunization campaign was launched by EPI that targeted 1.8 million children from 38 districts to prevent the further escalation in measles cases and it covered 98% of the child population between 6-59 months successfully [9]. Despite all these efforts made by the local and international organizations measles is still looming around. A huge resurgence has been witnessed where the global measles cases increased by 55% from 171,156 to 306291 during 2022-2023 [10]. As per a surveillance report by the CDC during the first half of 2023, approximately 10,549 measles cases were reported from Pakistan, making it the third major contributor to the measles global burden following India and Yemen, and during July-December 2024 the number of cases are still significant at 7,148 which brings Pakistan on the top of the list for the countries constantly battling with the measles disease burden [11].
In 2022-2023, a substantial measles outbreak in Pakistan necessitated a multifaceted study that employs epidemiological and molecular methodologies to elucidate the outbreak's complexities. This research explores comorbidities, complications, and molecular dynamics of the measles virus (MV), thereby advancing our comprehension of this critical public health issue. Moreover, the substitutional analysis of the circulating virus is an effort to dig deeper into the structural and functional variation in the measles virus and find out possible associations between the modifications in the virus and the severity of the disease during the current outbreak in Pakistan.
2. Methodology
2.1.Ethical Approval
The current research was approved by the Bio-Ethical Committee (BEC-FBS-QAU2023-520) at Quaid-i-Azam University Islamabad.
2.2. Sample population
Throat swabs were collected from patients (n = 183) admitted at Children’s Hospital measles ward, Pakistan Institute of Medical Sciences (PIMS) Islamabad, Pakistan during December 2022- December 2023. All subjects showed typical measles manifestations with symptoms such as fever, cough, rash, conjunctivitis, etc. The enrolled subjects were aged between 3 months and 9 years of age and were divided into four age groups i.e. infants (0-12 months), Toddlers (13-36 months), Early Childhood (37-72 months), and Middle childhood (73- 144 months). All the biological samples were collected after the written consent from the guardian/parent of the patients. Patient’s history and data including clinical, epidemiological, and vaccination status, were recorded from their hospital admission forms. Throat swab samples were transported to the Infectious Diseases and Molecular Pathology Laboratory (IDMPL), Quaid-I-Azam University Islamabad.
2.3. Statistical Analysis
Statistical analysis of patient’s clinical data was performed using SPSS-21 software. The frequency of symptoms and complications presented by the patients and duration of stay at the hospital among different age groups and both genders were analyzed using descriptive statistics. The normality test was carried out followed by the Kruskal Wallis test for comorbidities w.r.t., age groups. A p-value below 0.05 was considered significant. Post- hoc Mann-Whitney U test was used for different age groups and genders w.r.t., average stay at hospital and comorbidities.
2.4. Primer designing
The primers for the N gene were designed on the NCBI primer designing tool, primer blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/), by using reference sequence NC_001498.1. Forward primer 5’ATCCTGCTCTTGGACTGCAT’3 and reverse primer 5’TGGGAGTGGATGGTTGGTTG’3 was used for amplification of partial sequence of N gene including N-450 region that has been recommended by Global Measles and Rubella Laboratories Network (GMRLN) for Measles genotyping. The specifically designed primers anneal to a region spanning nucleotides 949-1794 in the MV genome, generating an amplicon of approximately 846 bp in size.
2.5. Viral RNA Extraction and synthesis of complementary DNA (cDNA)
by reverse transcription Viral RNA was extracted from throat swab samples of measles patients using WizPrep™ Total RNA Mini Kit by following the standard manufacturer’s protocol and the extraction was carried out in a designated extraction room with negative pressure and equipped with a biosafety cabinet Class II type B2. The complementary DNA was synthesized from extracted RNA using RevertAid first strand cDNA synthesis kit (Thermo Scientific, USA) and reverse primer that was designed for the N gene. The composition of the reaction mixture for reverse transcription of a total 20 μl reaction is given as follows; 5x reaction buffer 4 μl, 10mM dNTP 2 μl, specific primer 2 μl, Revert Aid transcriptase 1 μl, nuclease-free water 2.5 μl, RiboLock (RNase inhibitor) 0.5 μl, Template RNA 8 μl. Reaction mixture incubation was performed at 25 °C for 5 minutes and 42 °C for 60 min, followed by 45 °C for 30 min and a heat inactivation step (70 °C for 5 min).
2.6. PCR amplification of N gene (partial)
The amplification was carried out by using specifically designed and optimized primers for the conserved region of the measles N gene. The 20 μl of PCR mixture contained 10 μl HI Fidelity Phusion master mix (Thermo Scientific, USA) forward and reverse primers (2 μl), template cDNA (2 μl), and distilled water 6 μl. The cyclic condition of PCR was 5 minutes at 96°C, followed by 35 cycles with 1 cycle comprising 45 s at 96 °C, 30 s at 58 °C and 1 min at 72 °C with and final extension of 72°C for 10 min.
2.7. Visualization by gel electrophoresis and nucleotide sequencing
The amplified PCR products were run on agarose gel (2%) followed by visualization under UV trans illuminator. The targeted bands were excised from the gel and purified using the GeneJET Gel Extraction Kit (Thermo- Scientific, USA) following the standard protocol provided by the manufacturer. The eluted product (amplified product of N gene partial sequence) was stored at -20°C. The PCR product was sequenced via Sanger sequencing with the gene-specific forward primers. The deduced sequences obtained by Sanger sequencing were visualized by using the BioEdit software package [12].
2.8. Alignment and phylogenetic analysis of nucleotide sequences
The analysis of the obtained sequences was carried out utilizing genetic data resources for the measles virus N gene that were available at the National Center for Biotechnology Information (NCBI). Retrieved sequences were aligned at BioEdit, and MEGA-11 software was used for the construction of a phylogenetic tree [13] by using the Neighbors joining method, and the bootstrap values were determined by 1000 replicates in MEGA-11 [14]. The sequencing data for all the viral isolates were submitted to GenBank
2.9. Alignment and analysis of amino acid sequences
The nucleotide sequences were translated into amino acid sequences using the Expasy tool. The amino acid sequences were also aligned on MEGA-11 software, and then aligned via CLC sequence viewer (version 8) software with a reference sequence of WLY62623.1 reported from Russia
2.10. Prediction of Secondary and Tertiary Structures
The secondary structure of all isolates and reference proteins was predicted through the online server PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) (Accessed on 03, April 2024). The 2D Model of amino acid sequences was generated through the server by using PSI-BLAST and neural networking [15]. The tertiary structure of isolates was predicted through the Robetta server (https://robetta.bakerlab.org/) (Accessed on 03 April 2024). To construct the 3D structure of the protein, the server searches for an appropriate amino acid sequence template which is used for comparison modeling. If the amino acid of the template is missing the server utilizes de novo Rosetta fragment insertion approach [16]. For the prediction of 3D structures and identification of amino acid substitutions, Phymol software was utilized.
3. Results
In this study we investigated the population characteristics of 183 measles patients <9>
3.1. Population characteristics
Data of total 183 patients showed the mean age of the measles patients included in this study was 16.75 ± 1.6 months with a majority being male 52.45% and infants including children <12 xss=removed>
| Sr No. | Variables | Incidence in the studypopulation |
i. | Age | |
| i. Infant (0-12m) | 135 (73.77%) | |
| ii. Toddlers (13-36 m) | 29 (15.84%) | |
| iii. Early Childhood (37-72m) | 12 (6.55%) | |
| iv. Middle Childhood (72-144m) | 7 (3.82%) | |
| ii. | Sex | |
| i. Male | 96 (52.45%) | |
| ii. Female | 87 (47.5%) | |
| iii. | Comorbidities | |
| Jarco-Levin syndrome | 1 (0.54%) | |
| ASD with pulmonary hypertension | 1 (0.54%) | |
| Epilepsy | 2 (1.09%) | |
| Down syndrome | 3 (1.63%) | |
| Nephrocalcinosis | 1 (0.54%) | |
| Seizure Disorder with globaldelay | 1 (0.54%) | |
Complex congenital heart disease with situs inversus | 1 (0.54%), | |
| Splenomegaly | 1 (0.54%) | |
| Syndromic developmental delay | 2 (1.09%), | |
| Cerebral palsy | 4 (2.18%) | |
| Hypo calcemic fits | 1 (0.54%) | |
| Storage disorder | 1 (0.54%) | |
| Congenital diaphragmatic hernia febrile fits | 1 (0.54%) | |
| Anemia | 180 (98%) | |
| Hereditary spherocytosis | 1 (0.54%) | |
| Enteric fever | 1 (0.54%) | |
| Pancytopenia | 1 (0.54%) | |
| Meningitis | 1 (1.09%) | |
| GERD aspiration pneumonia | 1 (0.54%) | |
| iv. | Outcomes | |
| i. Discharged | 177 (96.72%) | |
| ii. Expired | 4 (2.18%) | |
| iii. LAMA | 2 (1.09%) | |
v. | Vaccination status | |
| i. Fullyvaccinated | 49 (26.77%) | |
| ii. Partially vaccinated. | 53 (28.96%) | |
| iii. Not vaccinated | 81 (44.26%) | |
| vi. | Antiviral therapy | |
| i. Given | 110 (60.10%) | |
| ii. Not given | 73 (39.8%) | |
| vii. | Average duration of hospitalization | 5.74 0.2 days |
| Association of hospital stay | ||
| Groups | P value | |
i. | Infants | 0.000 |
ii. | Toddlers | 0.000 |
iii. | Infant-Toddlers | 0.953 |
| Association of Symptoms | ||
| Pneumonia | ||
i. | Infants - Toddlers | 0.025 |
ii. | Infants - Middle childhood | 0.000 |
iii. | Toddlers - Middle childhood | 0.015 |
iv. | Early childhood- Middle childhood | 0.016 |
| Encephalitis | ||
i. | Infants - Toddlers | 0.005 |
ii. | Infants – Early childhood | 0.008 |
ASD: AtrialSeptal Defect, GERD: | Gastroesophageal RefluxDisease, LAMA: Left Admission without Medical Advice. | |
Table 1: Overview of the characteristics of the study populatio
The results showed a high incidence of complications among infants w.r.t., to age groups and among male measles patients. Rash, pneumonia, and conjunctivitis were the most prevalent complications followed by gastrointestinal (GI) symptoms, seizures, encephalitis, croup, and hepatitis (Figure 1).
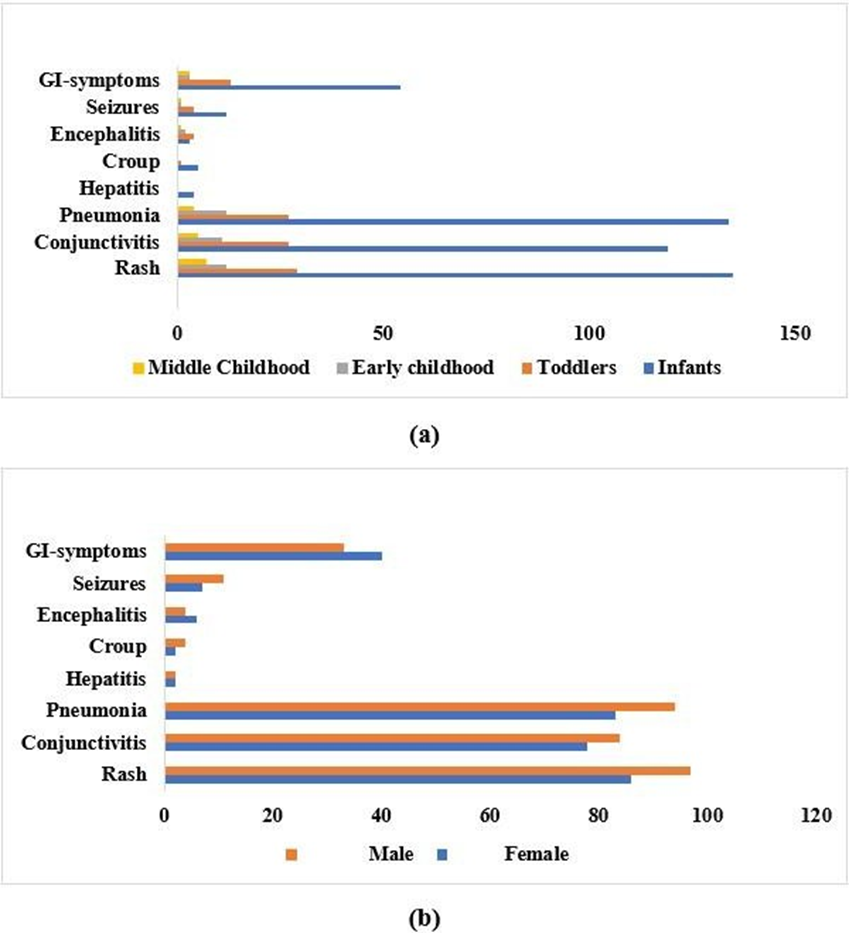
Figure 1: The incidence of complications in measles patients. (a) Presence of complications w.r.t.,
3.3. Results of statistical analysis
The duration of hospital stays varied significantly among different age groups. Specifically, the stay was significantly longer for infants (p = 0.000) and Toddlers (p = 0.000), but there was no significant difference between these two groups (p = 0.953). In contrast, the duration of hospital stay did not differ significantly between genders (p = 0.451).
Regarding the association of symptoms with different age groups, most of the symptoms including conjunctivitis, hepatitis, croup, seizures, and gastrointestinal symptoms showed no significant difference ( p>0.05 ). However, except pneumonia (p = 0.000) and encephalitis (p = 0.014) were significantly different among different age groups. Further group-wise comparison revealed that pneumonia was significantly different between, Infants- Toddlers (p = 0.025), Infants-Middle childhood (p = 0.000), Toddlers-Middle Childhood (p = 0.015), Early Childhood-Middle Childhood (p = 0.016). Similarly, Encephalitis was significantly different between Infants- Toddlers (p = 0.005), and Infants-Early Childhood (p = 0.008). Lastly, the analysis of symptoms with respect to gender showed no significant differences (p>0.05), indicating that symptoms prevailed equally among both genders.
3.4. Sequencing of positive measles virus sample
The amplified product was purified from gel and subjected to Sanger sequencing. The obtained sequences of MV N-gene were further processed and analyzed by using different bioinformatics tools
3.5. Phylogenetic analysis of partial nucleoprotein gene sequence of MVs
To evaluate the evolutionary history of our viral isolates we carried out phylogenetic analysis with the other closely related previously reported sequences by using the Neighbor-Joining method. The data for the previously reported measles virus isolates was retrieved from GenBank. The results of the evolutionary analysis revealed that our sequenced samples (QAUPAK1, QAUPAK2, QAUPAK3, QAUPAK4 isolates) were closely related to the measles virus reported from Russia and USA (accession number: OR290098 and OP314496) (Fig. 2). The sequences obtained from the viral isolates used in our current research were submitted to GenBank under the accession number OR622937, PP502160, PP502161, and PP502162.

Figure 2 Evolutionary analyses of measles virus from Pakistan (n = 4) with the reference sequence re
3.7. Substitutional analysis in amino acid sequence
We translated our nucleotide sequence into amino acid sequences using Expasy translate tool and aligned them with the CLC sequence viewer. Amino acid sequences (Isolate names: QAUPAK1 QAUPAK2, QAUPAK3, QAUPAK4) were aligned with reference sequence WLY62623 reported from Russia due to phylogenetic proximity. The alignment of our sequences did not show any abnormalities in the sequence such as gaps, insertions, or deletions, etc. The analysis of amino acid revealed close similarities with the measles virus sequences that had been reported from Russia previously. There were a total of 3 substitutions, identified by the amino acid sequence analysis of query sequences w.r.t., reference sequence. According to the reference sequence, QAUPAK1 isolate showed substitution at V394D, whereas S422G substitution was uniform for QAUPAK1, QAUPAK2, and QAUPAK3. Another substitution S487R was recorded in QAUPAK4. The details of substitutions have been presented in Table 2 and Figure 3.
| Sr no. | Amino acid position | Reference seq. WLY62623. 1 | Charge /polarit y | Query sequences | ||||
| Charge/polarity | QAUPAK1 | QAUPAK2 | QAUPAK3 | QAUPAK4 | ||||
| 1. | 394 | Valine | Unchar ged/non polar | -ve charged/polar | Aspartic acid | -- | -- | -- |
| 2. | 422 | Serine | Unchar ged/pol ar | Uncharged/nonpolar | Glycine | Glycine | Glycine | -- |
| 3. | 487 | Serine | Unchar ged/pol ar | +ve charged/polar | -- | -- | -- | Arginine |
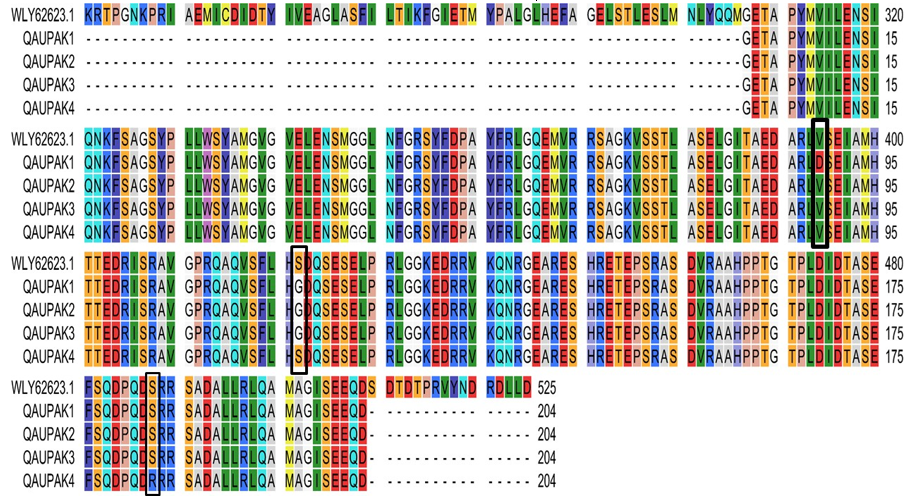
Figure 3: Amino acid sequence information of query sequences (QAUPAK1, QAUPAK2, QAUPAK3, QAUPAK4) w.
3.8.Secondary and Tertiary Structures Prediction
Online server PSIPRED was utilized for the investigation of secondary structures of all the isolates and reference proteins. The server provided information about the Beta strand, Alpha Helix, and Extended Coils as shown in Figure 4. The results illustrated that there is almost no difference present in the secondary structure. The percentage of Beta strands, Alpha Helix, and Extended Coils are given in Table 3. Moreover, the Robetta server was utilized for the tertiary structure of isolates and reference proteins. The tertiary structures of our isolates were compared with reference protein and amino acid substitutions were identified using Phymol. The 3D structure of isolates and reference protein along with amino acid substitutions are given in Figure 5.
| Sequences | Beta strand | Alpha helix | Extended coil |
| WLY62623.1 | 1.96% | 36.76% | 61.27% |
| QAUPAK1 | 2.94% | 36.76% | 60.29% |
| QAUPAK2 | 1.96% | 36.76% | 61.27% |
| QAUPAK3 | 1.96% | 36.76% | 61.27% |
| QAUPAK4 | 1.96% | 36.76% | 61.27% |
Table 3 Percentage of Beta strand, Alpha helix, and Extended coil in reference protein and all isola
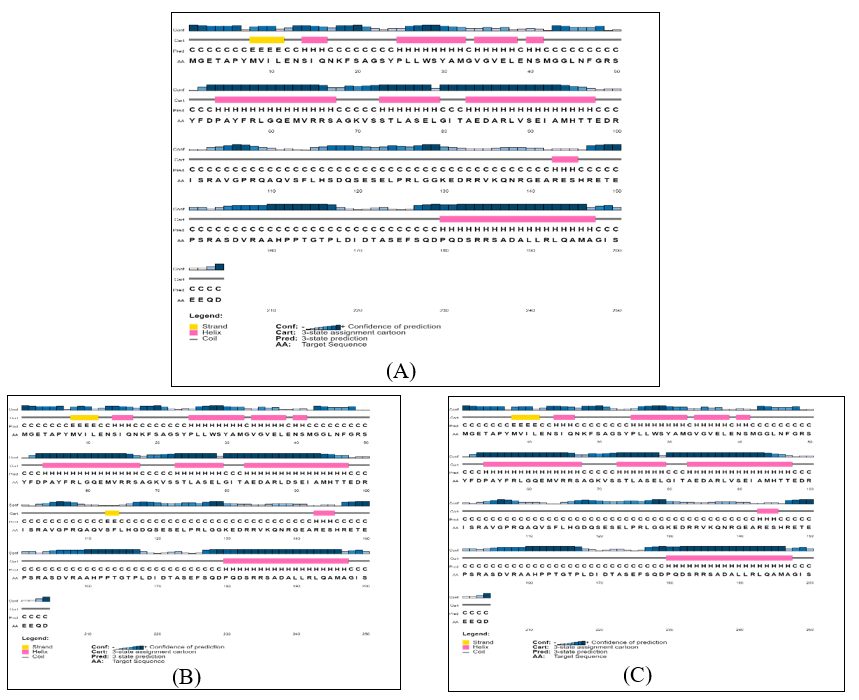
Figure 4: Secondary structure representation of all isolates and reference protein. (A), (B), (C), (

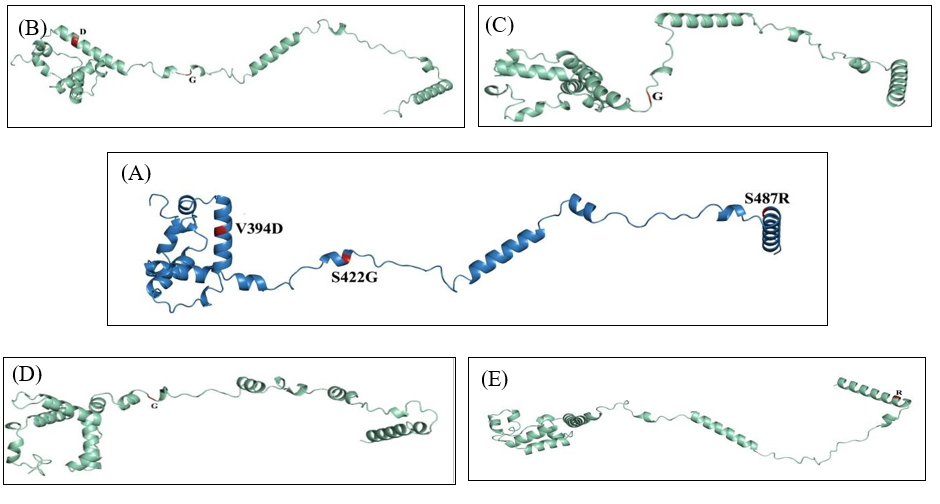
Figure 5: Tertiary structure representation of all isolates and reference protein. (A) Represents re
4. Discussion.
From November 2022 to April 2023, 6426 new measles cases were reported in Pakistan [17]. Measles usually gives rise to many different complications that increase inpatient hospitalization [18]. Our study explored illness severity and viral genetic characterization among hospitalized children during the 2022-2023 epidemic in Pakistan. It revealed a relatively high incidence (73.77%) of measles among infants (<12>
The phylogenetic analysis revealed that the isolates belong to the B3 genotype and were evolutionarily close to the viral isolates reported from the Russia and USA. Recent studies have identified the presence of B3 genotype in measles cases reported from both USA and European regions [24-25], implying genetic affinity and potential cross border transmission into Pakistan. Our results are consistent with the findings from past studies where the B3 genotype is the most prevalent worldwide and has been detected in the Pakistani region in various outbreaks during the past two decades [1, 26]. However, a few cases from other genotypes including H1 and D genotypes had also been reported in some regions during previous outbreaks [26].
Nucleoprotein is the most abundant, highly immunogenic structural protein that plays a crucial role in the regulation of replication, transcription, genome packaging and gene expression of morbilliviruses. [27-29]. It comprises 525 amino acids and contains two domains i.e. the NCORE (residues 1-400) interacts with the viral RNA and maintains nucleocapsid structure and the NTAIL (residues 401-525) that is a long intrinsically disordered domain that anchors the polymerase complex. Disordered NTAIL contains a molecular recognition element (MoRE) (residues 485-502) whose interaction with the C-terminal three-helix bundle domain XD of phosphoprotein (residues 459-507) leads to the recruitment of polymerase complex onto the nucleocapsid template [30]. Our study encompasses the analysis of 203 C-terminal residues (306-509) that form the anterior portion of the NCORE domain and a major (87.2%) portion of the NTAIL domain including molecular recognition element (MoRE). The substitutional analysis of 203 amino acids from the C-terminal of nucleoprotein showed a total of 3 substitutions concerning the reference sequence (WLY62623.1). In QAUPAK1 the V394D substitution was recorded in the terminal portion of NCORE domain. Valine is a medium-sized, hydrophobic, aliphatic, uncharged polar amino acid that prefers to be buried in protein hydrophobic cores. Valine is Cβ branched which means that unlike most of the other amino acids, they have two non-hydrogen substituents attached to its Cβ carbon which means that there is more bulkiness around the protein backbone and more restrictions in the conformation that the main chain can assume as a result valine is least likely to assume α helical structure. Valine has been substituted by Aspartic acid, which is a small, hydrophilic, negatively charged polar amino acid, and contrary to valine it prefers to be on the surface of protein [31]. Aspartic acid is frequently involved in the formation of salt bridges and its negative charge confers the ability to interact with the positively charged non-protein atoms such as zinc hence they are frequently involved in the protein active or binding sites. We can speculate that substitution with amino acids having opposite attributes can have a possible effect on the functioning of the NCORE domain and the possibility of altered interaction with the viral RNA and maintenance of nucleocapsid structure [32]. The second substitution S422G was seen uniformly across QAUPAK1, QAUPAK2, and QAUPAK3. Both serine and glycine are very small uncharged amino acids with the former being polar and later nonpolar. Serine is distinct from other amino acids with hydrogen as a side chain rather than carbon like all other amino acids. With this uniqueness in structure if a conserved serine changes into any other amino acid there can be a significant functional impact. Another substitution S487R was recorded that lies in the NTAIL region where serine was replaced by arginine which is a positively charged polar amino acid. Arginine mostly forms part of the active site and interacts with negatively charged proteins [31]. Due to diverse roles of N protein in Measles virus life cycle, we can speculate that the substations in the amino acid sequence of N gene would have impact on the structure and function of Measles virus [29].
The secondary structures were built de novo for viral isolates (QAUPAK1, QAUPAK2, QAUPAK3, QAUPAK4), and reference sequence (WLY62623) which showed a greater degree of similarity with some minor changes in the orientation of protein 3D structure. All the isolates formed a higher ratio of ≥ 60 of extended coil structure, 36.76% of α helix, and >1% of β strand. The variations in the protein structure can be due to the intrinsic disorders in the nucleoprotein of morbilliviruses or may be due to the amino acid substations observed in the study.
Measles in Pakistan remains a persistent challenge due to several contributing factors. While children younger than 9 months typically acquire immunity from maternal antibodies against measles, the high prevalence of the disease in this age group is alarming. Our study did not record the maternal medical or vaccination history, suggesting a need for future research focusing on women of childbearing age. Investigating post-COVID changes in maternal immunity could provide valuable insights into the prevalence of measles in young children. Furthermore, children are often admitted to hospitals at later stages of the disease when the viral load is declining, complicating molecular detection efforts. The simultaneous admission of multiple children from the same family underscores the necessity of raising awareness about measles infectiousness. Promoting early admission of measles patients could help prevent the spread of infection to healthy children. Additionally, our study focused on a single gene that exhibited significant substitutions and structural changes. Future studies should extend to other critical genes, exploring their mutational, structural, and functional characteristics. This could offer deeper insights into potential viral mutations that may be compromising the efficacy of current vaccines and treatments.
5.Conclusion
In conclusion, our study highlights the critical importance of integrating epidemiological and molecular approaches to explain the complexities of measles outbreaks, particularly among the concerning surge observed globally and especially in Pakistan between 2022 and 2023. By meticulously analyzing both the epidemiological data and molecular characteristics of the measles virus (MV), we have elucidated the severity of the disease among hospitalized children, the persistent circulation of the B3 genotype, and the significant variations in viral structure and amino acid substitutions, potentially influencing treatment effectiveness. Our findings emphasize the necessity for continuous surveillance, complete gene sequencing, and a deeper understanding of strain variations at both structural and functional levels to inform more efficacious treatment strategies and alleviate the burden on healthcare systems. This study serves as a crucial step towards combating measles outbreaks and offers valuable insights applicable to the management of future infectious disease challenges
6.Acknowledgements
We acknowledge the admitted patients, their guardians, and the PIMS staff for their cooperation during the sample collection.
Conflict of interest: The authors declare no competing interests.
Ethics Approval and Consent to Participate: This study was approved by the Bio-Ethical Committee (BEC-FBS- QAU2023-520) at Quaid-i-Azam University Islamabad. Participants provided informed consent to participate in the study.
Funding: No funds, grants, or other support was received.
Availability of Data and Material: The data will be available from the corresponding author upon reasonable request.
Permission to reproduce material from other sources: In this study, we didn't reproduce material from other sources
References
- aidi SS, Hameed A, Rana MS, Alam MM, Umair M, Aamir UB, Hussain M, Sharif S, Shaukat S, Angez M, Khurshid A. Identification of measles virus genotype B3 associated with outbreaks in Islamabad, Pakistan, 2013–2015. J Infect Public Health. 2018 Jul 1;11(4):540-5.
View at Publisher | View at Google Scholar - Laksono BM, de Vries RD, Verburgh RJ, Visser EG, de Jong A, Fraaij PL, Ruijs WL, Nieuwenhuijse DF, van den Ham HJ, Koopmans MP, van Zelm MC. Studies into the mechanism of measles-associated immune suppression during a measles outbreak in the Netherlands. Nat commun. 2018 Nov 23;9(1):1-0.
View at Publisher | View at Google Scholar - Nawaz A, Saeed A, Rahman S, Haider N. Hematological and Biochemical Assessment of Children Infected with Measles Virus: 2022 Outbreak in Pakistan: Measles’s Hematological and Biochemical Assessment. Proc Pak Acad Sci B Life and Enviro Sci. 2023 Jan 16;60(S):55-63.
View at Publisher | View at Google Scholar - Bai, P., Salik, M., Rais, H., Fawad, B., Sadar, S., & Saad, M. (2023). The spectrum of measles in COVID-19 pandemic ; An observational study in children . 30(08), 1009–1014.
View at Publisher | View at Google Scholar - Fisher DL, Defres S, Solomon T. Measles-induced encephalitis. QJM: An International Journal of Medicine.
View at Publisher | View at Google Scholar - 2015 Mar 1;108(3):177-82.
View at Publisher | View at Google Scholar - Garg RK, Suresh V, Suvirya S, Rizvi I, Kumar N, Pandey S. Clinical features, pathogenesis, pathology, neuroimaging, clinical course, and outcome of measles inclusion-body encephalitis: a systematic review of published case reports and case series. Neurol Sci. 2024 Mar 21:1-23.
View at Publisher | View at Google Scholar - Unicef. Pakistan to immunize more tham 90 million children against measles and rubella. Available from: https://www.unicef.org/pakistan/press-releases/pakistan-immunize-more-90-million-children-against-measles- and-rubella [Accessed 12 November 2023].
View at Publisher | View at Google Scholar - World Health Organization. Measles-rubella campaign undertaken to prevent disease outbreak in Pakistan. Available from: https://www.emro.who.int/pak/pakistan-news/measles-rubella-campaign-undertaken-to-prevent-disease-outbreaks-in-pakistan.html [Accessed 20 November 2023]
View at Publisher | View at Google Scholar - Durrheim DN, Murray P, Turner N. Resurgent global measles: A threat to Australia, New Zealand, and Pacific Island Countries. Journal of Paediatrics and Child Health. 2024 Mar 1.
View at Publisher | View at Google Scholar - Center for Disease Control and Prevention. Global Measles Outbreaks. Available from: https://www.cdc.gov/globalhealth/measles/data/global-measles-outbreaks.html
View at Publisher | View at Google Scholar - Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. InNucleic acids symposium series 1999 Jan 1;41(41):95-98.
View at Publisher | View at Google Scholar - Tamura K, Stecher G, Kumar S. MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol.
View at Publisher | View at Google Scholar - 2021 Jul 1;38(7):3022-7.
View at Publisher | View at Google Scholar - Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. evolution. 1985 Jul 1;39(4):783-91.
View at Publisher | View at Google Scholar - McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000 Apr 1;16(4):404-5.
View at Publisher | View at Google Scholar - Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic acids research. 2004 Jul 1;32 Suppl 2 S526-31.
View at Publisher | View at Google Scholar - Muzzamil M, Fatima F, Baloch R, Hashmi S, Malikzai A. Under-5 children in Pakistan are disproportionately affected by the alarming increase in measles cases. IJS Global Health. 2023 Sep 1;6(5):e0334.
View at Publisher | View at Google Scholar - Chovatiya R, Silverberg JI. Inpatient morbidity and mortality of measles in the United States. PLoS One. 2020 Apr 28;15(4):e0231329.
View at Publisher | View at Google Scholar - Federal
View at Publisher | View at Google Scholar - Jensen MR, Communie G, Ribeiro Jr EA, Martinez N, Desfosses A, Salmon L, Mollica L, Gabel F, Jamin M, Longhi S, Ruigrok RW. Intrinsic disorder in measles virus nucleocapsids. Proc Natl Acad Sci USA. 2011 Jun 14;108(24):9839-44.
View at Publisher | View at Google Scholar - Sindhu TG, Geeta MG, Krishnakumar P, Sabitha S, Ajina KK. Clinical profile of measles in children with special reference to infants. Trop Doct. 2019 Jan;49(1):20-3.
View at Publisher | View at Google Scholar - Shah SJ, Dero AA, Hayat N, Khan MJ, Ahmad S, Ahmad H. Frequency of complications of measles in hospitalized children of Pakistan. J Pharm Negat Results. 2022 Dec 1:3757-62.
View at Publisher | View at Google Scholar - Diwan MN, Samad S, Mushtaq R, Aamir A, Allahuddin Z, Ullah I, Ullah Afridi R, Ambreen A, Khan A, Ehsan N, Ehsan Khattak Z. Measles induced encephalitis: recent interventions to overcome the obstacles encountered in the management amidst the COVID-19 pandemic. Diseases. 2022 Nov 17;10(4):104.
View at Publisher | View at Google Scholar - Probert WS, Glenn-Finer R, Espinosa A, Yen C, Stockman L, Harriman K, Hacker JK. Molecular epidemiology of measles in California, United States—2019. The Journal of Infectious Diseases. 2021 Sep 15;224(6):1015-23.
View at Publisher | View at Google Scholar - Kalinichenko S, Melentyeva K, Toryanik I, Zvereva N, Antusheva T, Buriachenko S. Genetic monitoring of endemic measles virus circulation in European countries. Annals of Mechnikov's Institute. 2020(2):57-70.
View at Publisher | View at Google Scholar - Zaidi SS, Hameed A, Ali N, Umair M, Alam MM, Rana MS, Sharif S, Aamir UB, Shaukat S, Angez M, Khurshid A measles outbreak in Sindh, Pakistan caused by a genotype B3 virus. Arch virol. 2017 Aug 12;162:3603- 3610.
View at Publisher | View at Google Scholar - Baloi B. In silico epitope prediction and 3D model analysis of peste des petits ruminants virus nucleoprotein (PPRV N). bioRxiv. 2016 Dec 19:095505.
View at Publisher | View at Google Scholar - Lund GA, Tyrrell DL, Bradley RD, Scraba DG. The molecular length of measles virus RNA and the structural organization of measles nucleocapsids. J Gen Virol. 1984 Sep;65(9):1535-42.
View at Publisher | View at Google Scholar - Parks CL, Lerch RA, Walpita P, Wang HP, Sidhu MS, Udem SA. Comparison of predicted amino acid sequences of measles virus strains in the Edmonston vaccine lineage. Journal of virology. 2001 Jan 15;75(2):910-20.
View at Publisher | View at Google Scholar - Bankamp B, Horikami SM, Thompson PD, Huber M, Billeter M, Moyer SA. Domains of the measles virus N protein required for binding to P protein and self-assembly. Virology. 1996 Feb 1;216(1):272-7.
View at Publisher | View at Google Scholar - Betts MJ, Russell RB. Amino acid properties and consequences of substitutions. Bioinform Geneticists. 2003 Feb 21:289-316.
View at Publisher | View at Google Scholar - Karlin D, Longhi S, Canard B. Substitution of two residues in the measles virus nucleoprotein results in an impaired self-association. Virology. 2002 Oct 25;302(2):420-32.
View at Publisher | View at Google Scholar -
View at Publisher | View at Google Scholar
