

Review Article | DOI: https://doi.org/10.31579/2835-8325/073
Diagnosis, treatment and monitoring of Adrenocortical carcinoma
- Tabouri Sarah 1*
- Belkralladi Houria 2
- CHELEF Sidi Mohamed Amine 3
- BENCHOUK Jesia Asma 4
- Maaref Nawel 5
1Department of Medical Oncology, Center for the fight against cancer of Sidi Bel Abbes. Taleb Morad Faculty of Medicine – Djillali Liabes University- Algeria.
2Department of pathology – University Hospital Hassani Abdelkader - Taleb Morad Faculty of Medicine – Djillali Liabes University- Algeria.
3Department of Urology - University Hospital Hassani Abdelkader - Taleb Morad Faculty of Medicine – Djillali Liabes University- Algeria.
4Breast and pelvic Surgery Department – Center for the fight against cancer of Sidi Bel Abbes. Taleb Morad Faculty of Medicine – Djillali Liabes University- Algeria.
*Corresponding Author: Tabouri Sarah, Department of Medical Oncology, Center for the fight against cancer of Sidi Bel Abbes. Taleb Morad Faculty of Medicine – Djillali Liabes University- Algeria.
Citation: Tabouri Sarah, Belkralladi Houria, Chelef Mohammed Amine, Benchouk Jazia Esmaa, et al (2024), Diagnosis, treatment and monitoring of Adrenocortical carcinoma, Clinical Research and Clinical Reports, 4(3); DOI: 10.31579/2835-8325/073
Copyright: © 2024, Tabouri Sarah. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 06 April 2024 | Accepted: 20 April 2024 | Published: 26 April 2024
Keywords: adrenocortical carcinoma; guidelines; diagnosis; treatment; follow-up
Abstract
The adrenocortical carcinoma (ACC) is a primary malignant tumor developed from the adrenal cortex.
The annual incidence of this tumor is estimated between 0.5 and 2 cases per million inhabitants. They are most often sporadic tumors. Generally, ACCs are revealed by hormonal symptoms or symptoms due to an abdominal mass. Radiologically, the size of the tumor can be predictive of the malignancy of an adrenal mass. Beyond 6 cm, the proportion of malignant tumors is 25%, while it is less than 2% for masses less than 4 cm. The CT scan has a sensitivity and specificity of 71 and 98%, respectively, and abdominal MRI does not provide additional diagnostic information compared to the CT scan (its sensitivity and specificity are lower: 78 and 87% respectively in tissue characterization). Treatment is based on surgery. Chemotherapy is indicated in case of R2 resection or as an adjuvant if high risk.
We will report the case of a young patient operated on for adrenocortical carcinoma, the postoperative CT scan revealed an early recurrence which was revealed to be a textiloma on MRI. Through this case we will describe the different clinical, radiological, therapeutic and prognostic aspects of these rare tumors, and synthesize the current recommendations for the diagnostic, therapeutic and surveillance management of adrenocortical carcinoma.
We will report the case of a young patient operated on for adrenocortical carcinoma, the post-operative CT scan revealed an early recurrence which turned out to be a textiloma on MRI. Through this case, we will describe the different clinical, radiological, therapeutic and prognostic aspects of these rare tumors, and summarize the current recommendations for the diagnosis, treatment and monitoring of ACC.
Introduction
Malignant adrenal tumors (MATs) are either primary tumors affecting the cortex of the gland (adrenocortical carcinoma or malignant adrenocortical tumor), or the adrenal medulla (malignant pheochromocytoma), or adrenal metastases from another cancer, or lymphomas [1].
Adrenocortical carcinoma (ACC) is a rare tumor, with an annual incidence of 0.5 to 2 per million inhabitants. It is more frequent in women (ratio 1.5). It can occur at any age but has two peaks of onset, in the first decade and between 40 and 50 years [1]. ACCs are most often sporadic and are rarely associated with other endocrine neoplasms (MEN type 1, Beckwith-Widman syndrome, Li-Fraumeni syndrome) [2].
Case Presentation
We report the case of a 28-year-old young female patient with Neurofibromatosis type I, also known as Recklinghausen disease, with a surgical history of tumorectomy for thoracic neurofibromatosis nodules during her adolescence. Clinically, the patient presents with hirsutism and Cushing's syndrome.
The onset of the disease dates back to six months before diagnosis with the onset of diffuse abdominal pain with bloating. An abdominal ultrasound was performed, revealing a mass in the left hypochondrium, which appeared to be related to her neurofibromatosis. Due to the persistence of symptoms, an abdominal CT scan was performed, this time highlighting a large, roughly oval mass occupying the left hypochondrium and the left flank, well-defined, of retroperitoneal origin given the anterior pancreatic displacement, developing at the level of the left adrenal lodge with displacement of the left kidney downwards without signs of kidney infiltration; this mass measures 186/174/145mm with predominantly tissue density with heterogeneous enhancement delimiting an area of central necrosis. This mass compresses the left renal vein causing perirenal and perilesional venous collateral circulation. This appearance suggested a primary or secondary adrenal tumor lesion. In view of this appearance, the patient was operated on, the procedure performed was a monobloc resection of the tumor mass.
Anatomopathological study of the surgical specimen revealed the following macroscopic findings a large encapsulated circumscribed tumor measures 22/15/10 cm. The cut surface shows a yellowish-tan variegated appearance, with many intratumorally nodules. Areas of necrosis and hemorrhage are seen (Figure 1).
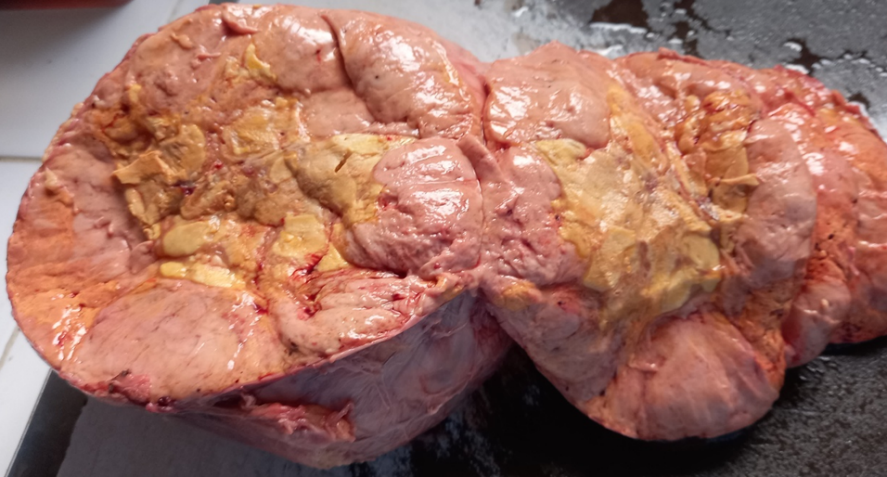
Figure 1: Macroscopic Appearance of the Surgical Specimen after Formalin Fixation
(Photo credit: Prof. BELKRALLADI Houria)
Microscopic Examination (figure 2 and 3): The tumor exhibits a predominantly encapsulated architecture, characterized by thick fibrous capsules surrounding the neoplastic tissue. The internal structure demonstrates a variety of growth patterns, including solid areas, broad trabeculae, and large nests of cells. Necrosis is a frequent finding within the tumor mass. Evidence of both capsular and vascular invasion is present, indicating aggressive behavior. The mitotic index is elevated, with 4 mitoses identified per 50 high-power fields. According to the Weiss scoring system, the tumor receives a score of 7. Evaluation using the Bisceglia criteria reveals the presence of two major criteria, further supporting the diagnosis.
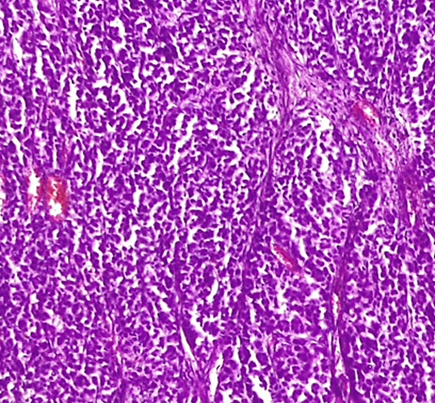
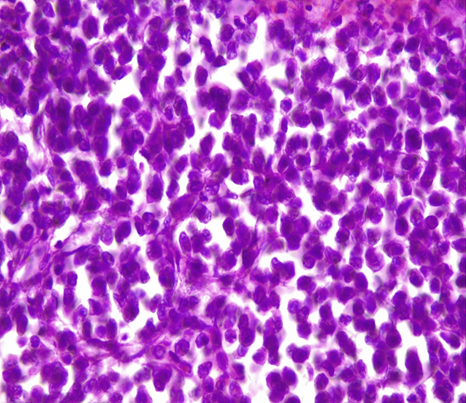
Figure 2: Microscopic Examination
Left: HE x 10 large trabeculae and nests/ Right: HE x 20 large nests
(Photo credit: Prof. BELKRALLADI Houria)
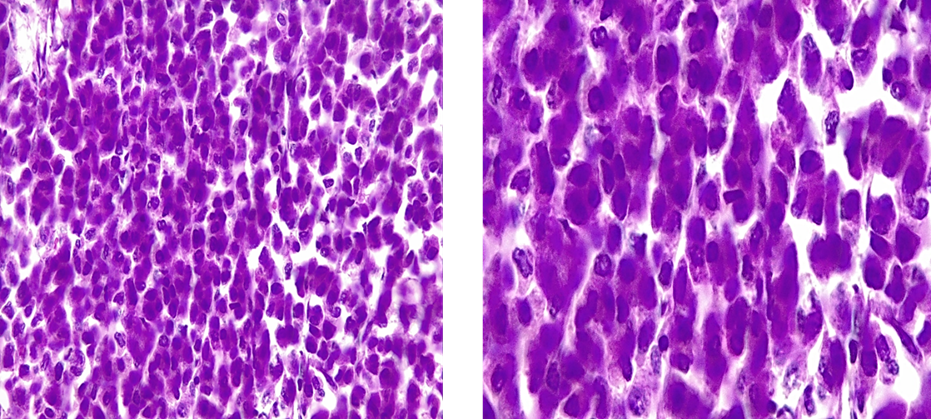
Figure 3: Oncocytic cells tumor > 75%
Left: HE x20 / Right: HEx 40 Nuclear grade 3 (Furhman)
(Photo credit: Prof. BELKRALLADI Houria)
A thoraco-abdominopelvic CT scan performed three weeks post-operatively revealed a well-defined, polylobulated retroperitoneal mass located in the left anteromedial para-renal space. The mass measured 53 x 25 mm with an extension of 56 mm, displacing the kidney without visualization of the ipsilateral adrenal gland. Heterogeneous enhancement with areas of liquefaction was observed, along with significant infiltration of the surrounding fat. Hormonal workup showed no abnormalities.
Following a multidisciplinary team discussion, the patient was initiated on a chemotherapy regimen consisting of Doxorubicin at 20mg/m² on days 1 and 8, Cisplatin at 40mg/m² on days 2 and 3, and Etoposide at 100mg/m² from days 5 to 7, with a cycle repeated every 28 days. Granulocyte colony-stimulating factor (G-CSF) was administered concurrently.
After six cycles of chemotherapy, an evaluation CT scan demonstrated a slight reduction in tumor size, maintaining the same relationship with adjacent organs. A subsequent abdominal MRI revealed a well-encapsulated, non-infiltrative lesion in the upper pole of the left kidney with homogeneous signal intensity and no dilatation of the collecting system, likely representing a textiloma (surgical sponge).
Following a multidisciplinary team discussion, a second surgical intervention was recommended, but the patient declined. After a 12-month interval, a repeat abdominal MRI showed perfect stability of the previously described lesion. The patient remains under surveillance, currently 30 months from the initial diagnosis.
However, several issues hindered the management of our patient, first and foremost the lack of experienced radiologists in our center, which initially led us astray. Indeed, there was confusion between a textiloma and an early tumor recurrence on the CT scan, although the CT scan is the examination of choice for diagnosing recurrence. The other difficulty we faced was the non-availability of Mitotane in our hospital, so the patient received chemotherapy without this major treatment for the management of adrenocortical carcinomas.
Discussion
Adrenocortical carcinoma (ACC) presents with a spectrum of clinical manifestations. The most frequent presentations include hormonal hypersecretion (40-74%) and symptoms related to tumor mass effect (40-60%). Hormonal hypersecretion can manifest as Cushing's syndrome (30%), virilization (20%), feminization (10%), or mixed hormonal secretion (35%) [2]. Incidental discovery during imaging studies accounts for 10-20% of cases, while paraneoplastic syndromes are rare [3]. Features suggestive of malignancy include rapid symptom onset, large tumor size, lumbar pain, fever, anorexia, and weight loss. Genetic evaluation is essential in ACC management, as 10% of cases arise within hereditary cancer syndromes (Table1) [4]. Several syndromes are associated with an increased risk of ACC, including [4]:
- Li-Fraumeni syndrome: Characterized by TP53 mutations, this syndrome predisposes individuais to various cancers, including ACC, often at an early age. It is present in 50-80% of children and 3-5% of adults with ACC.
- Lynch syndrome: Caused by mutations in mismatch repair (MMR) genes (MSH2, MSH6, MLH1, PMS2), this syndrome increases the risk of colorectal, endometrial, ovarian, and other cancers, including ACC. Approximately 3% of adult ACC patients carry germline MMR gene mutations.
- Beckwith-Wiedemann syndrome: Associated with chromosome 11 abnormalities, this syndrome is characterized by macroglossia, omphalocele, and an increased risk of developing tumors, including ACC.
- Neurofibromatosis type 1 (NF1): Individuals with NF1, caused by NF1 gene mutations, present with neurofibromas and flat, light brown spots on the skin , and a higher risk of developing various tumors, including ACC. The young age of onset in the presented case is consistent with the presence of NF1.
A comprehensive clinical evaluation is crucial in the management of suspected ACC [5-8]. The assessment should focus on identifying signs and symptoms suggestive of steroid hormone hypersecretion, including Cushing's syndrome, hyperandrogenism in females, hypoestrogenism in males, and hypertension with hypokalemia. Additionally, evaluation for a tumor syndrome is essential, encompassing the presence of a palpable mass, pain, or compressive symptoms related to the primary tumor or potential distant metastases.
Further key elements of the clinical evaluation include: assessment of symptom chronology, rapid onset of symptoms may indicate a more aggressive disease course; evaluation for genetic predisposition, a thorough family history and personal history of other malignancies should be obtained to identify potential hereditary cancer syndromes associated with ACC; exclusion of differential diagnoses is another conditions that can mimic ACC, such as metastatic cancer or pheochromocytoma, should be ruled out through appropriate investigations, and assessment of comorbidities and performance status: Comorbidities and overall health status significantly impact prognosis and guide treatment decisions [8].
Syndrom | Gene | Prevalence in ACC patients |
Li-Fraumeni | TP 53 | 3-5% (adults) 50-80% (pediatric) |
Lynch | MSH2, MSH6, MLH1, PMS2 | 3% (adults) |
Multiple endocrine neoplasia 1 | MEN1 | 2?ults) |
Familial adenomatous polyposis | APC | <1> |
Beckwith Wiedemann | H19 IGF2 | <1> |
Carney complex | PRKAR1A | <1> |
Neurofibromatosis type 1 | NF1 | <1> |
Table 1: Hereditary Syndromes Associated with Adrenocortical Carcinoma [4]
A comprehensive metabolic panel is essential to assess blood glucose, complete blood count, potassium levels, and renal and hepatic function, particularly to evaluate the potential impact of hypercortisolism. Specific investigations should include assessment for steroid hormone hypersecretion, including cortisol, aldosterone, testosterone in females, estradiol in males, and dehydroepiandrosterone sulfate (DHEA-S) [6-8]. Measurement of plasma and/or urinary metanephrines and normetanephrines is necessary to rule out pheochromocytoma [5].
Urinary steroid profiling using mass spectrometry can also be employed [6]. This test can demonstrate the secretion of steroid metabolites, aiding in the diagnosis of ACC in over 80% of cases. Additionally, it provides prognostic information, as tumors producing glucocorticoids are associated with a higher mortality risk, and it helps inform the risk of post-operative adrenal insufficiency [6].
Computed tomography (CT) is the primary imaging modality for visualizing adrenocortical carcinomas. These tumors typically appear as heterogeneous adrenal masses with a spontaneous density greater than 10 Hounsfield units (HU) on unenhanced scans. CT has a sensitivity of 97% but a moderate specificity of around 52% for ACC [7]. Characteristic features include areas of necrosis and heterogeneous enhancement. While tumor size is often greater than four centimeters, smaller ACCs can also be detected. Thoraco-abdominopelvic CT is essential for evaluating locoregional invasion (including adipose tissue, abdominal lymph nodes, vena cava, adrenal vein, and renal vein) and distant metastases (commonly affecting the liver, lungs, abdominal lymph nodes, mediastinal and supraclavicular lymph nodes, and less frequently, bones and peritoneum).
Abdominal (and hepatic) MRI can be used as an alternative to CT in cases of contraindication or as a complementary modality to better assess locoregional and lymph node involvement, particularly when there is suspicion of metastasis or hepatic invasion [2].
PET-FDG should be routinely performed before surgery for suspected localized or oligometastatic ACC [3].
Percutaneous biopsy is not recommended as a first-line diagnostic tool for suspected ACC, particularly if the primary tumor is hormonally active and/or resectable, due to the risk of tumor dissemination. Adrenal biopsy does not always reliably differentiate ACC from atypical adrenocortical adenoma [8]. However, biopsy may be considered in select cases, such as when the diagnosis is uncertain for a non-secreting tumor or a tumor of non-adrenocortical origin (e.g., adrenal metastasis from an unknown primary, lymphoma, or sarcoma). Biopsy may also be considered for molecular analysis or to assess tumor grade (Ki-67%) for potential neoadjuvant chemotherapy [8]. When performed, biopsy should be done using a retroperitoneal approach with a large-bore 16G (or even 14G) coaxial needle under CT guidance to minimize the risk of dissemination [9].
Immunohistochemical analysis is essential to confirm the adrenocortical origin of the tumor. Steroidogenic factor 1 (SF1) is the most specific marker for adrenocortical tissue. Other less specific markers, such as Melan A, Inhibin alpha, Calretinin, Synaptophysin, and Chromogranin A, can be used in conjunction with SF1 but should not be relied upon in isolation, as they are typically negative in ACC [4].
For localized adrenocortical tumors in adults, histopathological evaluation and prognostication rely on assessing architectural, cytological, invasive, and proliferative features. These features are combined into multiparametric scoring systems, along with Ki-67 proliferation index assessment. Several validated scoring systems exist:
Weiss score: The Weiss score [10] is the most widely used system and is applied to the most common types of adrenocortical tumors.
Lin-Weiss-Bisceglia score: This system is specifically designed for oncocytic adrenocortical neoplasms, where the Weiss score is not applicable and may overestimate malignant potential [11].
Other scoring systems, such as the Helsinki score and reticulin network evaluation, are currently under investigation [12].
The Ki-67 proliferation index is a crucial prognostic biomarker in ACC and plays a role in determining the need for adjuvant therapy.
The pathology report for adrenocortical tumors should encompass the following crucial elements:
- Specimen integrity: A thorough assessment of the surgical specimen, noting any capsular rupture or fragmentation.
- Tumor size and weight: Precise measurement of the tumor in three dimensions (length, width, and depth) and its weight.
- Weiss score: Evaluation and reporting of all nine components of the Weiss scoring system.
- Mitotic count: Accurate quantification of mitotic figures per 10 mm², specifically within 50 high-power fields at 400x magnification with a field diameter of 0.2 mm². This count is essential for differentiating between low-grade and high-grade adrenocortical carcinoma (ACC).
- Ki-67 proliferation index: Determination and reporting of the percentage of tumor cells exhibiting positive staining for the Ki-67 proliferation marker.
- Resection status (R): Precise description of the surgical margin status, including the minimal microscopic margin distance.
- Lymph node assessment: The number of lymph nodes examined and the number of lymph nodes found to contain metastatic involvement (N).
- Distant metastasis (M): Documentation of the presence or absence of distant metastases, including any resected metastatic lesions.
- pTNM staging: Assignment of the appropriate pathological tumor-node-metastasis (pTNM) stage according to the American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) eighth edition TNM classification system.
- Cryopreservation status: Indication of whether any tumor tissue has been cryopreserved for potential future analyses.
Prognostic classifications for ACC are based on the TNM staging system and the GRAS factors (Grade, Resection status, Age, and Symptoms). Table 2 presents the risk stratification for recurrence in patients with resected, localized ACC, while Table 3 summarizes the prognostic stratification for overall survival in patients with advanced ACC[8].
Risk Group | Features | Recurrence Risk |
Low Risk | Stage 1-2, R0 resection, Ki-67 ≤ 10%, N0 | 10% |
Intermediate Risk | Stage 1-2, R0 resection, Ki-67 10-20%, N0 | 30-50% |
High Risk | Stage 3 and/or R1 resection and/or Ki-67 > 20% | 50-70% |
Table 2: Risk Stratification for Recurrence in Resected, Localized ACC [8]
Prognostic Group | Features | Estimated 5-Year Survival |
Intermediate Prognosis | Unresectable Stage 3 or N1 R0 resection of primary, Ki-67 < 20%, Weiss score ≤ 6, absence of symptoms, age < 50 years | 15-50% |
Poor Prognosis | Stage 4 with ≥ 3 tumor-involved organs Unresected primary and/or Ki 67 > 20% and/or Weiss score > 6, symptomatic ACC (hormonal hypersecretion or tumor syndrome), age ≥ 50 years | 0-15% |
Table 3: Prognostic Stratification for Overall Survival in Advanced ACC [8]
The mainstay of treatment for localized, resectable ACC is surgery. This approach is applicable to ACC cases where complete surgical resection is feasible, encompassing stages 1, 2, and select stage 3 tumors. The goal of surgery is to achieve an R0 resection, which is attainable in over 95% of cases. The risk of mortality associated with surgery is less than 5%, and the morbidity risk typically does not impede the initiation of adjuvant mitotane therapy within six weeks post-operatively [13].
Surgery remains the sole potentially curative treatment for localized ACC. Ideally, it should be performed within four weeks of diagnosis, adhering to oncological surgical principles, with the primary aim of achieving a complete resection without tumor violation. Data from expert networks and registries indicate that only 56-85% of initial resections achieve R0 status [14]. The resection should encompass, at minimum, the adrenal gland and periadrenal fat en bloc. In some cases, achieving a complete (R0) resection may necessitate sacrificing adjacent organs (kidney, vena cava, spleen, liver, pancreas, and/or stomach) to obtain a monobloc resection with clear margins and without tumor violation [15]. Preservation of the ipsilateral kidney is desirable whenever possible to maintain adequate renal function, particularly in cases where systemic therapy is anticipated [16].
Even in the absence of suspicious lymphadenopathy, lymph node dissection should be routinely performed for staging purposes. This dissection should include, at minimum, the periadrenal and perirenal lymph nodes, as well as those at the renal hilum. Any suspicious lymph nodes identified on CT, TEP-FDG, or intraoperatively should also be resected [17]. Perioperative hydrocortisone replacement is necessary for all patients with cortisol-secreting tumors.
Radiotherapy may be considered as an alternative to surgery in the rare cases of non-metastatic ACC that are deemed inoperable due to significant comorbidities precluding surgery or general anesthesia [3]. However, data on the efficacy of radiotherapy in this setting are limited, and such decisions should be made within a multidisciplinary team setting.
The role of adjuvant radiotherapy following surgery, with or without mitotane, particularly in cases of Rx, R1, or R2 resections, remains a subject of debate [2].
Adjuvant therapy with mitotane is routinely recommended after surgical resection for patients with intermediate or high risk of recurrence (as outlined in Table 2). For patients with a low risk of relapse, surveillance is generally the preferred approach.
Mitotane is a medication with a long half-life, dose-limiting toxicity, and a narrow therapeutic window. The current recommendation is to achieve a mitotane blood level between 14-20 mg/L or the highest tolerated dose [18].
Most experts suggest that adjuvant mitotane therapy should be continued for at least two years due to the high frequency of recurrences during this period. Beyond two years, continuation of mitotane may be considered for patients with a high risk of relapse, good tolerance to the medication, and mitotane levels within the therapeutic range [2]. The total duration of adjuvant therapy generally does not exceed five years. It is important to note that increased FDG uptake in the contralateral adrenal gland has been reported, particularly during the initial phase of mitotane treatment [19].
The management of recurrent ACC is individualized based on prognostic factors, including the disease-free interval, type of recurrence (locoregional or metastatic), resectability, and surgical risk. Recurrences are classified as either early (disease-free interval less than six months) or late (disease-free interval greater than twelve months) based on the timing relative to initial surgery [2,19].
For early recurrences, systemic therapy with EDP-M (Etoposide, Doxorubicin, Cisplatin, and Mitotane) is recommended as first-line treatment, rather than locoregional therapy [4].
In cases of late recurrence, repeat surgery is considered the optimal treatment for resectable, localized lesions, particularly if the initial surgery achieved R0 resection [2]. Surgery should be performed as early as possible [19]. Focal therapy may also be considered if complete ablation is feasible. Adjuvant mitotane therapy is recommended as soon as possible following surgery for late recurrences [18].
Beyond first-line therapy, there is no established standard of care. Various protocols have been utilized, including Gemcitabine and Capecitabine, 5-Fluorouracil plus Streptozotocin, Temozolomide, and Cabozantinib [20-22].
The prognosis for patients with metastatic ACC is generally poor, with a five-year overall survival rate of less than 15%. However, there is significant heterogeneity in disease behavior. The median survival for aggressive metastatic disease is approximately one year, while it is two years for cases with moderate tumor burden (two or fewer metastatic organs). Unresectable stage III disease has a median survival of five years [23]. Prolonged survival beyond five years can occur, particularly in patients with "intermediate" prognosis and/or those who respond well to mitotane or chemotherapy [23-25]. Figure 4 summarizes the treatment strategy for ACC [2].
Following complete resection, a rigorous surveillance protocol is recommended, involving clinical, hormonal, and imaging assessments (thoraco-abdominopelvic CT or 18F-FDG PET) every 3 months for the initial 2 years, followed by every 3 to 6 months for the next 3 years [1]. Beyond 5 years, continued surveillance may be considered on a case-by-case basis. For advanced ACC, the surveillance protocol should be tailored based on prognostic factors, expected treatment efficacy and toxicity, and available alternative treatment options. Regular hormonal evaluation is recommended for all patients [1].
In cases of advanced disease, closer monitoring is recommended, with follow-up assessments every one to three months, depending on tumor aggressiveness and the chosen treatment approach. Typically, patients with metastatic ACC undergo comprehensive biological and radiological evaluations every two to three months during active treatment [8].
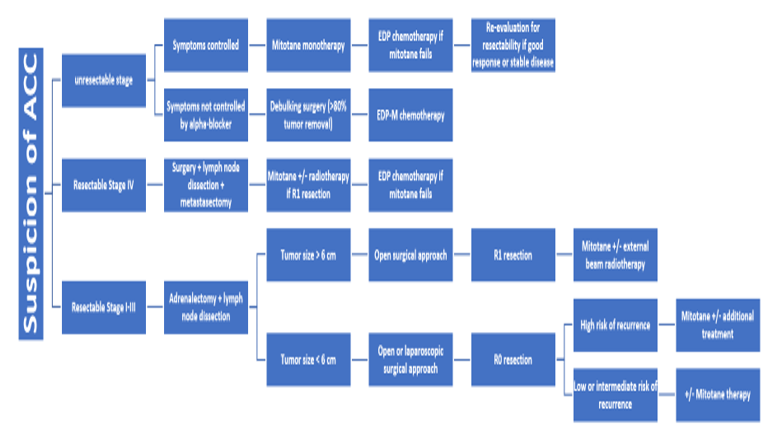
Figure 4: ACC Management Algorithm
EDP: Etoposide-Doxorubicine-Cisplatine; EDP-M: Etoposide-Doxorubicine-Cisplatine et Mitotane; R0: No residual tumor; R1: Microscopic residual tumor; Ki67: Immunohistochemical expression of Ki-67.
Conclusion
Adrenocortical carcinoma is a rare and aggressive malignancy with a heterogeneous clinical presentation and disease course. Early diagnosis and complete surgical resection offer the best chance for cure in localized disease. Multidisciplinary management is crucial, incorporating surgery, adjuvant mitotane therapy, and consideration of radiotherapy and systemic chemotherapy in select cases. Prognosis is influenced by tumor stage, grade, and molecular features. Ongoing research efforts are focused on improving prognostication, identifying novel therapeutic targets, and developing more effective treatment strategies to improve outcomes for patients with ACC.
References
- Fassnacht M, Dekkers O, Else T, Baudin E, Berruti A, De Krijger RR, et al. (2018). European Society of endocrinology clinical practice guidelines on the management of adrenocortical carcinoma in adults, in collaboration with The European Network For the study of adrenal tumors. Eur J Endocrinol.
View at Publisher | View at Google Scholar - Savoie P-H, et al. (2018). Recommandations françaises du Comité de Cancérologie de l’AFU — Actualisation 2018-2020 tumeur de la surrénale. Prog Urol (2018),
View at Publisher | View at Google Scholar - Fassnacht M, Dekkers O, Else T, Baudin E, Berruti A, Krijger R, et al. (2018). European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol, 179 :1-46.
View at Publisher | View at Google Scholar - Mete O, Erickson LA, Juhlin CC, de Krijger RR, Sasano H, Volante M, Papotti MG. (2022). (Overview of the 2022 WHO Classification of Adrenal Cortical Tumors. Endocr Pathol, 33(1):155-196.
View at Publisher | View at Google Scholar - Fassnacht M, Dekkers O, Else T, Baudin E, Berruti A, Krijger R, et al. (2018). European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol, 179: 1–46.
View at Publisher | View at Google Scholar - Arlt W, Biehl M, Taylor AE, Hahner S, Libé R, Hughes BA, et al. (2011). Urine steroid metabolomics as a biomarker tool for detecting malignancy in adrenal tumors. J Clin Endocrinol Metab, 96 :3775-3784.
View at Publisher | View at Google Scholar - Thomas AJ, Habra MA, Bhosale PR, Qayyum AA, Ahmed K, Vicens R, et al. (2018). Interobserver agreement in distinguishing large adrenal adenomas and adrenocortical carcinomas on computed tomography. Abdom Radiol N Y, 43 :3101–3108.
View at Publisher | View at Google Scholar - Rossella Libé, Magalie Haissaguerre, Karine Renaudin, Matthieu Faron, Myriam Decaussin-Petrucci, Fréderic Deschamps, Anne-Paule Gimenez-Roqueplo, Eric Mirallie, Thibaut Murez, François Pattou, Laurence Rocher, David Taïeb, Pierre Henri Savoie, Antoine Tabarin, Jérôme Bertherat, Eric Baudin, Christelle de la Fouchardière. 20232). Recommandations conjointes du réseau National ENDOCAN-COMETE, de l'Association francophone de chirurgie endocrinienne et de la Société française d'urologie pour la prise en charge du carcinome corticosurrénalien. Bull Cancer, 110:707-730.
View at Publisher | View at Google Scholar - Van Houdt WJ, Schrijver AM, Cohen-Hallaleh RB, Memos N, Fotiadis N, Smith MJ, et al. (2017). Needle tract seeding following core biopsies in retroperitoneal sarcoma. Eur J Surg Oncol, 43:1740-1745.
View at Publisher | View at Google Scholar - Weiss LM, Medeiros LJ, Vickery Jr AL. (1989). Patho-logic features of prognostic significance in adrenocortical carcinoma. Am J Surg Pathol, 13:202-206.
View at Publisher | View at Google Scholar - Bisceglia M, Ludovico O, Di Mattia A, Ben- Dor D, Sandbank J, Pasquinelli G, et al. (2004). Adrenocortical oncocytic tumors: report of 10 cases and review of the literature. Int J Surg Pathol, 12 :231-243.
View at Publisher | View at Google Scholar - Duregon E, Cappellesso R, Maffeis V, Zaggia B, Ventura L, Berruti A, et al. (2017). Validation of the prognostic role of the
View at Publisher | View at Google Scholar - Yip L, Duh QY, Wachtel H, Jimenez C, Sturgeon C, Lee C, et al. (2022). American Associa-tion of Endocrine Surgeons guidelines for adrenalectomy: executive summary. JAMA Surg, 157(10) :870-877.
View at Publisher | View at Google Scholar - Elhassan YS, Altieri B, Berhane S, Cosentini D, Calabrese A, Haissaguerre M, et al. (2021). S-GRAS score for prognostic classification of adrenocortical carcinoma: an international, multicenter ENSAT study. Eur J Endocrinol, 186:25-36.
View at Publisher | View at Google Scholar - Mirallié E, Blanchard C, Caillard C, Rodien P, Briet C, Mucci S, et al. (2019). Adrenocortical carci-noma: impact of surgical treatment. Ann Endocrinol (Paris), 80(5–6):308-313.
View at Publisher | View at Google Scholar - Calcatera NA, Hsiung-Wang C, Suss NR, Winchester DJ, Moo-Young TA, Prinz RA. (2018). Minimally invasive adrenalectomy for adre- nocortical carcinoma: five-year trends and predictors of conversion. World J Surg, 42:473-481.
View at Publisher | View at Google Scholar - Panjwani S, Moore MD, Gray KD, Finnerty BM, Beninato T, Brunaud L, et al. (2017). The impact of nodal dissection on staging in adrenocor- tical carcinoma. Ann Surg Oncol, 24:3617-3623.
View at Publisher | View at Google Scholar - Baudin E. (2015). Endocrine Tumor Board of Gustave Roussy. Adrenocortical carcinoma. Endocrinol Metab Clin North Am, 44:411-434.
View at Publisher | View at Google Scholar - Fassnacht M, Dekkers O, Else T, Baudin E, Berruti A, De KrijgerRR, et al. (2018). European Society of endocrinology clinical practice guidelines on the management of adrenocortical carcinoma inadults, in collaboration with The European Network For thestudy of adrenal tumors. Eur J Endocrinol 2018.
View at Publisher | View at Google Scholar - Sperone P, Ferrero A, Daffara F, Priola A, Zaggia B, Volante M, et al. (2010). Gemcitabine plus metronomic 5-fluorouracil or capecitabine as a second-/third-line chemotherapy in advanced adrenocortical carcinoma: a multi-center phase II study. Endocr Relat Cancer, 17:445-453.
View at Publisher | View at Google Scholar - Cosentini D, Badalamenti G, Grisanti S, Basile V, Rapa I, Cerri S, et al. (2019). Activity and safety of temozolomide in advanced adrenocortical carcinoma patients. Eur J Endocrinol, 181:681-689.
View at Publisher | View at Google Scholar - Kroiss M, Megerle F, Kurlbaum M, Zimmer- mann S, Wendler J, Jimenez C, et al. (2020). Objec- tive response and prolonged disease control of advanced adrenocortical carcinoma with cabozantinib. J Clin Endocrinol Metab, 105:1461-1468.
View at Publisher | View at Google Scholar - Gonzalez RJ, Tamm EP, Ng C, Phan AT, Vassilopoulou-Sellin R, Perrier ND, et al. (2007). Response to mitotane predicts outcome in patients with recurrent adrenal cortical carcinoma. Surgery, 142:867-875.
View at Publisher | View at Google Scholar - Malandrino P, Al Ghuzlan A, Castaing M, Young J, Caillou B, Travagli JP, et al. (2010). Prog-nostic markers of survival after combined mitotane- and platinum-based chemother-apy in metastatic adrenocortical carcinoma. Endocr Relat Cancer, 17:797-807.
View at Publisher | View at Google Scholar - Vezzosi D, Do Cao C, Hescot S, Bertherat J, Haissaguerre M, Bongard V, et al. (2018). Time until partial response in metastatic adrenocortical carcinoma long-term survivors. Horm Cancer, 9:62-69.
View at Publisher | View at Google Scholar
