

Research Article | DOI: https://doi.org/10.31579/2835-835X/001
Design of Potential Inhibitors of Urokinase Using in Silico Approach
1 Master Student in Pharmaceutical Chemistry and Drug Control Department, Faculty of Pharmacy,Tishreen University, Lattakia, Syria.
2 Professor in Pharmaceutical Chemistry Department, Faculty of Pharmacy, Tishreen University, Lattakia, Syria.
*Corresponding Author: Faten Alchab, Professor in Pharmaceutical Chemistry Department, Faculty of Pharmacy, Tishreen University, Lattakia, Syria.
Citation: Faten Alchab, Omar Rihawi. (2022) Design of Potential Inhibitors of Urokinase Using in Silico Approach. Clinical Trials and Case Studies.1(1); DOI:10.31579/2835-835X/001
Copyright: © 2022 Faten Alchab, This is an open-access artic le distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 26 September 2022 | Accepted: 10 October 2022 | Published: 20 October 2022
Keywords: urokinase; molecular modeling; docking; upa; cancer
Abstract
Urokinase-type plasminogen activator (uPA) plays an important role in the regulation of diverse physiological and pathological processes. Elevated uPA expression is associated with cancer progression, metastasis, and shortened survival in patients, whereas suppression of proteolytic activity of uPA leads to evident decrease of metastasis. Therefore, uPA has been considered as a promising molecular target for development of anticancer drugs. The aim of this study was to find effective and selective inhibitors of uPA using molecular modeling study. A library of chemical compounds derived from Naphtalin and Theophylline cores was then docked within the active site of the uPA. The binding affinity and the binding positions of the suggested compounds with the active site of the previously prepared uPA enzyme were determined. The study led to three effective and selective inhibitors of the uPA enzyme; one compound derived from the Naphtalin core -Naph 15- and two compounds derived from the Theophylline core -Theo17 and Theo 18-. The study was completed by chemical synthesis of Theo17 molecule.
Introduction
Proteolytic enzymes (proteases) comprise a family of enzymes which hydrolyse protein or peptide substrates in the generalized process of intracellular protein degradation, a process essential for the normal functioning of all cells [1]. Proteolytic processes are necessary for normal physiological functions in the body, the same enzyme system for these functions is also used by the cancer cells for their growth and spread. These enzymes are produced by the tumor cells or cells surrounding them and can degrade the basement membrane and extracellular matrix (ECM), [2]. Degradation of the surrounding connective tissue is considered a necessary step to allow malignant cells to locally invade, to enter the lymphatic or blood circulation and to metastasize [3]. Proteases include many groups, one of the most important groups is serine proteases. Urokinase-type plasminogen activator (uPA), the trypsin like serine protease is strongly associated with tumor cells and is implicated in a large number of malignancies, including cancers of the breast, lung, bladder, stomach, cervix, kidney, and brain, and high levels of urokinase have been correlated with poor patient prognosis [4]. Thus, there is great clinical interest in the development of potent and bioavailable inhibitors of urokinase that can serve as therapeutic agents for the treatment of cancer [5]. The uPA is a 53 kDa multidomain glycoprotein of 411 residues, glycosylated at Asn302, synthesized and secreted as a single-chain enzymogen (pro-uPA or sc-uPA); once released in extracellular environs, the sc-uPA is exposed to the action of proteases which may generate enzymatically active or inactive high-molecular-weight forms of uPA (HMW-uPA) [6]. Plasmin, cathepsin B and L, kallikrein, trypsin or thermolysin cleave the sc-uPA peptide bond K158-I159 converting the proenzyme in the active disulfide bridge-linked two-chain form (tc- uPA) [7]. Of the two chains, the N-terminal (A-chain) contains the kringle domain and the epidermal growth factor (EGF)-like domain, the latter responsible for the binding to uPAR, whereas the C terminal (B-chain) is composed of two subdomains formed by six -strands folded in an antiparallel manner and connected by twist or helical regions. The active site is located at the interface between the two subdomains, and consists of the catalytic triad of His57, Asp102 and Ser195 residues [8]. The tc-uPA has restricted substrate specificity with plasminogen as main substrate. Plasmin is the primary activator of sc-uPA and is in turn activated by tc-uPA, thus enhancing its own production. Such phenomenon, referred as “reciprocal zymogen activation”, occurs much more efficiently when the sc-uPA is associated with its receptor uPAR. As a consequence, the active uPA generation is concentrated in the pericellular area, where it represents an effective and rapid source of plasmin during cell migration and invasion under physiological or pathological conditions [9]. There are reports of nonpeptidic, reversible inhibitors of uPA from as far back as the late 1950’s. These small molecule inhibitors include benzamidines, phenylguanidines, acylguanidines and bisbenzamidines [10]. The best of these early uPA inhibitors have µM potencies and poor selectivity. Several novel uPA inhibitors with nM potency and selectivity towards uPA include benzothiopheneamidines [11] (Eisai), 5-thiomethylthiopheneamidine [12], Naphthylamidines [13] (Abbott), and isoquinolynylguanidines [14] (Pfizer). In the past few years, a number of novel small molecule uPA inhibitors have been proposed. However, among these inhibitors, only WX-UK1 (WILEX, Munich, Germany) entered clinical development, showing a against human uPA of 0.6 M. Since WX-UK1 is not absorbed orally, more recently, Wilex developed an oral prodrug, WX-671, for the systemic delivery of the active WX-UK1. This prodrug is currently under evaluation in two independent studies of phase II clinical trials in combination with classical cytotoxic treatments to estimate its efficiency [15]. The crystal structure of uPA catalytic domain displays a trypsin-like topology in which the Asp189 is retained, conferring to the S1 site an affinity for positively charged Arg and Lys residues [16]. Therefore, the majority of synthetic uPA inhibitors, conceived so far, share a common structural feature consisting of a mono- or biaromatic moiety substituted with an amidino or guanidino function, acting as arginine mimetic. However, a strong limitation in the choice of feasible compounds is represented by the necessity to inhibit uPA without affecting the activity of other trypsin-like serine proteases, and especially tPA and plasmin, essential for the fibrinolytic processes [17]. In the development of a clinical agent, selectivity for the target protein is important for reducing the potential for harmful side-effects. This is particularly true for the trypsin-like family of proteases that have been implicated in a number of highly regulated processes. The S1β pocket of bovine trypsin is different from that of urokinase primarily because of an asparagine substitution for Lys143 [18]; filling this site could result in a change of the specificity profile. Occuping S1β more fully, results in a loss of potency for trypsin while maintaining high potency for urokinase. Hence, binding at S1β appears to exploit structural differences between urokinase and trypsin, thus has resulted in potent and specific urokinase inhibitors [19]. Though, both the design and synthesis of a vast number of selective uPA inhibitors, there is still an inability to find critical chemical features of uPA inhibitors. This leads to discover new inhibitors by studying new molecules through molecular modeling and synthesizing them.
Methods and materials:
According to the importance of the inhibition of uPA enzyme and the crucial role of molecular modeling at this time in designing new inhibitors for various kinds of enzymes, the molecular modeling has been used in this research to design new inhibitors of uPA enzyme using Discovery Studio 2016 (DS) program and protein Data Bank (PDB), a huge bank which contains a big number of crystalline proteins and macromolecules with or without compounds attached to them.
Data set:
When preparing a set of data, all the available crystalline forms of uPA were collected from the PDB. A crystalline form of the enzyme in complex with an inhibitor was chosen for the study (pdb code: 1owd). Different amino acid residues in the active site are important for binding, such as the catalytic triad residues Ser195, Asp102, His57 and the S1 pocket residues like Asp189, Gly218 and Arg 217. Asp189 is located at the bottom of UPA’s S1 pocket. The negative charge of Asp189 is 2 important because it stabilizes amino acids with positively charged side chains which found in the substrate Plasminogen.The crystalline form of the uPA protein 1owd was associated with an inhibitor derived from the 2- Naphtamidine core. The shape is highly accurate R = 2.3A° and the Root Mean Square Deviation was less than 2 [18].
Preparation of the crystalline form of the uPA protein:
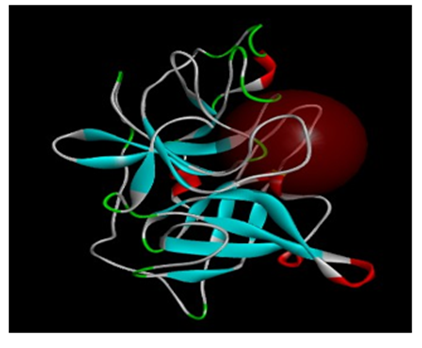
The protein structure which was used in the study (1OWD) was prepared. For docking, a CHARMm force field was applied and the hydrogen atoms were added. The global system was then minimized to reduce its energy without affecting the remains of the protein’s structure (as heavy atoms). After minimization, a 9.5 Angstrom sphere around the binding site was determined in order to study the association of the new inhibitors inside (Figure 1).
Set a library of new chemical compounds:
Depending on the nature of the cores commonly used in the design of uPA inhibitors, which showed good IC50 values, two cores were proposed to develop uPA inhibitors. The first core (Naphtalin) provides the appropriate dimensions for the ligand in order to fit within the active site. The second chosen core was Theophylline which is generally available with low price. After drawing and designing the compounds derived from these two cores, the study of the docking process of these compounds was carried out within the active site of the uPA enzyme.
Docking studies:
The docking process between the protein 1OWD and the compounds drawn from the cores was carried out using the CDocker process in DS program. The validation of the docking method was first investigated by comparing the standard deviation of the basic inhibitor with the deviation placement according to the CDocker method provided, where the standard deviation RMSD must be less than 2A°. This was followed by a docking procedure between 1OWD and the drawn compounds according to the protocol followed in the program.
After the completion of the docking process, the binding mode was studied and the binding affinity, which is expressed as a score function, was measured as the value of -CDocker energy. In other words, the binding of the CDocker method is indicated by the negative value of the CDocker energy.
Results and discussion:
As shown in Table1 and Table2, several potential substituents were included for the Naphtalin and Theophylline core. Many of the chemical substituents, including hydrogen and alkyl groups, were analyzed and their affinity was determined based on two basic principles. The first was the expression of the correlation between position, type of substituents and the binding affinity. The second was the best binding with the most important amino acids such as Ser195, Asp102, His57 and the S1 pocket residues like Asp189, Gly218 and Arg217. The Selection of the best compound was dependent on two parameters, the first was the score function and the second was the ability of binding with the important amino acids in the active site of uPA. In addition to that, the uPA enzyme has an additional extra hydrophobic pocket within the S1 pocket, it is called S1β which can be exploited. This can add an extra feature for the new inhibitor of the uPA enzyme [19].
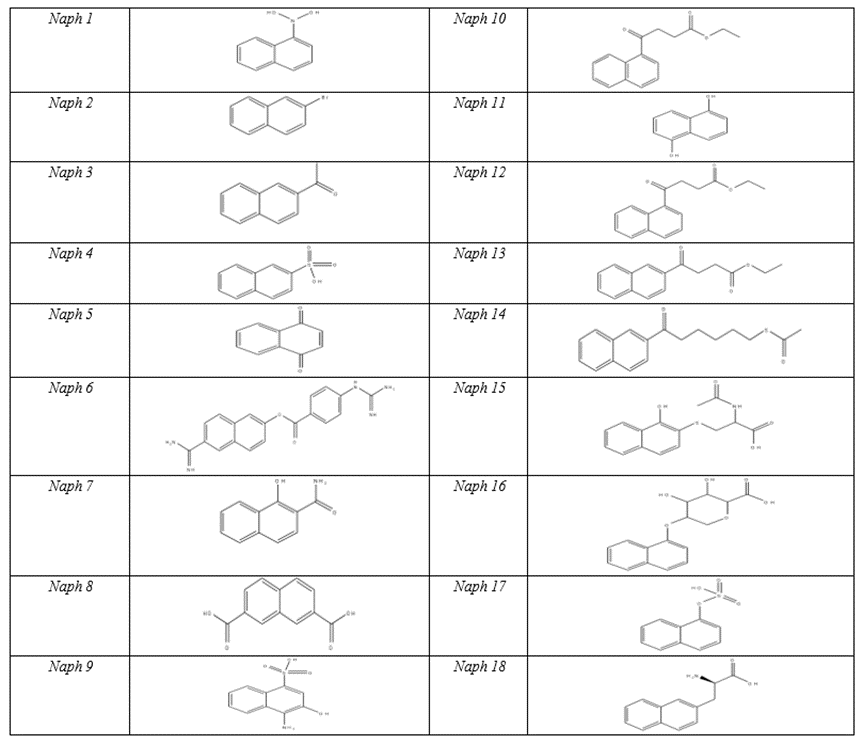
Regarding the Naphtalin core, many kinds of substituents were added in order to carry out a detailed study of the potential inhibitors within the active pocket based on the principles mentioned above. The results were discussed in detail. Table3 shows (-CDocker) energy values for the designed compounds.
It has been noted that the compounds like Naph1 and Naph2, which have small substituents do not have high binding affinity with uPA. However, when the substituents carry polar functional groups such as NH2, OH, the affinity of these compounds toward the enzyme increases, and that is because the polar functional groups form important hydrogen bonds with many amino acid residues such as Cys191,Ser195 and Gly218.
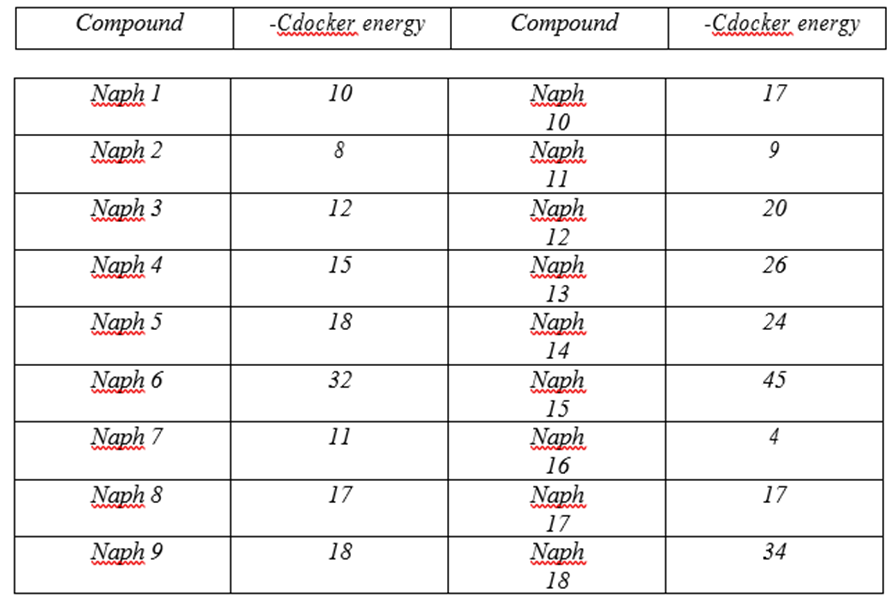
It turns out that it is important to add long side chains on C1 and C2 of Naphtalin with polar functional group content,in order to make interactions with more amino acid residuesinside the active site of the enzyme and that can be seen in the case of the compounds Naph10 and Naph14.These compounds bind to the S1pocket and especially make interactions with many residues such as Gln192,Cys191and His93.
By looking at the functional groups carried on the side chains of Naphtalin, we find that the carbonyl group plays an important role in the binding affinity and that can be clearly seen by looking at Naph15. The value of (–Cdocker) energy of Naph15 increases to 45, because of the carbonyl groups, which bind to many amino acid residues within the S1-pocket, like the salt bridge with Arg217 and the Pi-Stacking bonds between the two aromatic cycles of Naphtalin and Cys191 at the S1β Pocket. Occupying S1β, as mentioned above, results in a loss of potency for trypsin while maintaining high potency for urokinase. Hence, binding at S1β results in potent and specific urokinaseinhibitors.
On the other hand, we recognize the importance of Arg217.Arg217 is situated at the S1 pocket near Asp189,which is locatedat the bottom of S1pocket,and gives Urokinase and all tripsin-like serine proteases the difference in specificity among other serine proteases.Asp217 forms asalt bridge with the carboxyl group of Naph15 as well as with the carboxyl group of Naph18,and that resultsin an increase of thesecompounds’ binding affinity, in ordr to actually interactin a stabilizing manner, which will create a stable effect.
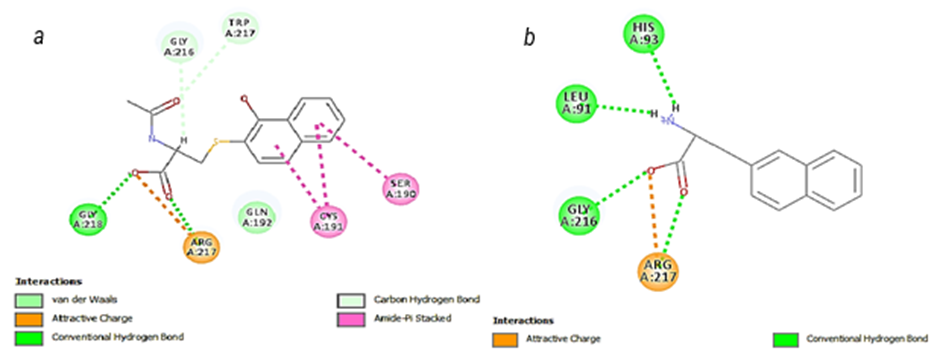
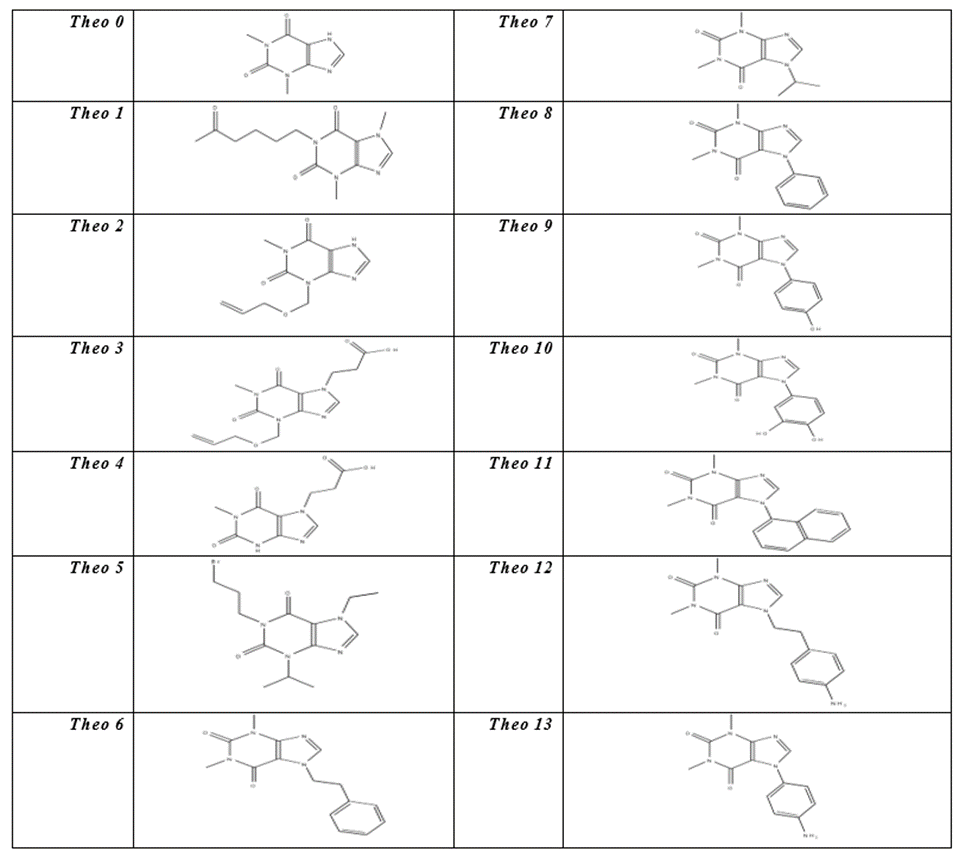
The core of Theophylline differs from the Naphtalin’s by the number of replaceable substituents. Theophylline’s core has a single adjustable substituent on N7. However, there is an efficient route to Theophylline derivatives, selectively substituted at the 1-, 3-, or 7- positions by the Cyclocondensation of a suitably protected aminoimidazole with methyl-2- phenylthioethyl carbamate, followed by oxidation of sulfur to the sulfone [20]. Like the compounds derived from the Naphtalin core, the results of (–Cdocker) energy of the compounds derived from Theophylline core were presented in Table4 due to the importance of these values in detecting the affinity of these compounds for the active site of uPA enzyme.
Compound | -Cdocker energy | Compound | -Cdocker energy | |||
Theo 0 | 27.7 | Theo10 | 16.5 | |||
Theo 1 | 36 | Theo11 | 0.5 - | |||
Theo 2 | 22 | Theo12 | 35 | |||
Theo 3 | 37 | Theo13 | 14 | |||
Theo 4 | 42.3 | Theo14 | 32 | |||
Theo 5 | 33 | Theo15 | 40 | |||
Theo 6 | 32 | Theo16 | 45 | |||
Theo 7 | 27 | Theo17 | 42 | |||
Theo 8 | 9 | Theo18 | 48 | |||
Theo 9 | 13 | |||||
Table 4: C Docker energy values of the compounds derived from Theophylline core
By looking at the docking results, we see that Theo0 interacts actually well with the active site of uPA by forming hydrogen bonds with the residues Arg217 and Gly218 at the S1 pocket, but it lacks the high binding affinity. It appears that the (–Cdocker) energy value of Theo0 is only 22.7.
It has been observed that the long side chain on the N1 site that carries polar functional groups contributes significantly to a high binding affinity, as is the case with Theo1, where the carbonyl group of Theo1 forms a hydrogen bond with Gln192 at the S1β pocket, in addition to other bonds with Gly218 and Arg217 at the bottom of S1 pocket.
On the other hand we see that the substituents on the N3 site do not result in high binding affinity. On the contrary, despite all the interactions with many important amino acid residues such as Ser195 at the S1 pocket and Gly218 at the S1β pocket, the (-Cdocker) energy of Theo1 is low, if we compare Theo1 with the core Theophylline.
By looking at Theo3, we notice the important effect of the side chain which carries a carboxyl group on the binding affinity with the enzyme as the (-Cdocker) energy increases to 37, because of the salt bridge with Arg217 and the hydrogen bonds with Gly216 and Gly218 within S1 pocket.
When we remove the side chain on the N3 site from Theo3, we will see that (–Cdocker) energy increases to 42.3 as is the case with Theo4. This asserts that the substitution on N3 site is not important.
So we focused on the N7 site of Theophylline and added side chains, which carry polar functional groups like the case of Theo5 and Theo6. As we expected, the binding affinity increased because (–Cdocker) energy of Theo5 and Theo6 reached the values 40 and 45 respectively. Moreover, an important hydrogen bond has been formed between Theo15 and Ser195, which is one of the catalytic triad residues. This hydrogen bond blocks the location at the catalytic triad and therefore, there will be no more catalysis.
By adding hydrophobic substituents on N7 site of Theophylline, we see that the binding affinity does not increase, which is the case with Theo8, Theo9, Theo10, Theo11 and Theo13. However, if we add polar functional groups to the aromatic cycle, the (–Cdocker) energy will increase slightly as is the case with Theo9 and Theo10.
If we concentrate on the nature of the substituents on the N7 site, we will recognize the importance of the side chains which carry polar functional groups, like in Theo4, Theo15 and Theo16. The (–Cdocker) energy values were 42.3, 40 and 45 respectively.
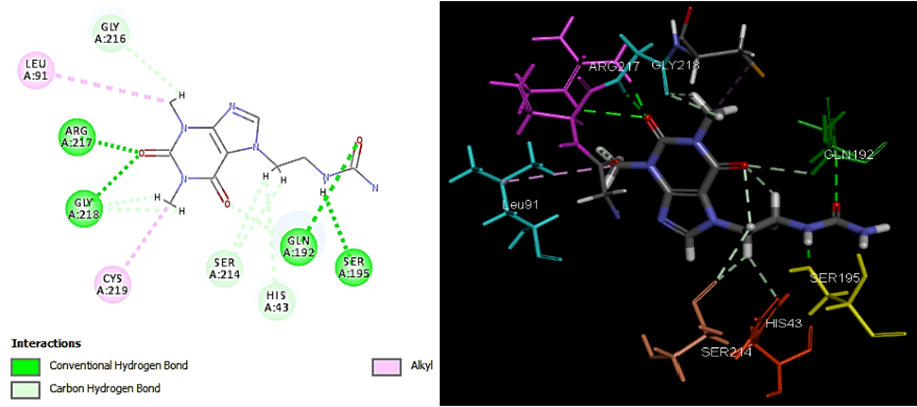
Through studying the compounds Theo17 and Theo18, a significant increase in the values of (-CDocker) energy was noticed. Besides, we see, in the case of Theo17, the very important hydrogen bond with Ser195 at the catalytic triad location, which blocks Ser195. Such localization of Theo17 would interfere with the ability of uPA to recognize its substrates and inhibit enzyme activity [21]. Moreover, there are other hydrogen bonds with Gln192 at S1β pocket and with Arg217 and Gly218 at the bottom of S1 pocket. As a result, the –Cdocker energy of Theo17 increases to 45.7. Theo18 in turn showes the highest –Cdocker energy among Theophylline compounds. We also see with Theo18 the unique hydrogen bond with Asp189. This amino acid residue gives all T specificity.
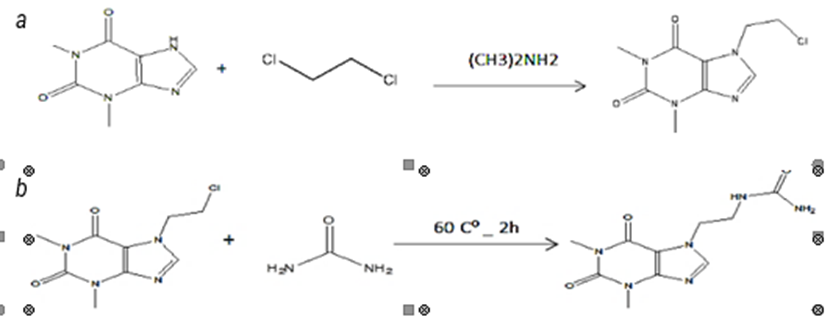
Theophylline derivatives like Theo17 and Theo18 can be synthesized, first by N-alkylation of the core unit with an alkyl group such as dichloroethane in presence of strong base according to caffeine synthesis process (figure 5) [22]. And then, N-alkylation between urea and the intermediate compound can be achieved to get Theo17 [23], or we can get Theo18 using tartaric acid with the same intermediate compound in the presence of an appropriate base [24].
Conclusion:
Molecular modeling was performed on uPA. Docking study using Naphtalin and theophylline cores led to identify different hits which had high affinity to the uPA active pocket, In addition to that, these compounds formed bonds with important amino acids such as Ser195,Cys191 and Asp189
References
- MANTLE D, PREEDY VR. (2002) Adverse and beneficial functions of proteolytic enzymes in skeletal muscle. Adverse Drug React. Toxicol Rev 21: 31–49.
View at Publisher | View at Google Scholar - WANG Y. (2001) The role and regulation of urokinase-type plasminogen activator receptor gene expression in cancer invasion and metastasis. Med Res Rev 21: 146–170.
View at Publisher | View at Google Scholar - DEL ROSSO M, FIBBI G, PUCCI M, D’ALESSIO S, DEL ROSSO Aet al. (2002) Multiple pathways of cell invasion are regulated by multiple families of serine proteases. Clin Exp Metastasis 19: 193–207.
View at Publisher | View at Google Scholar - Andreasen, P. A.; Kjoller, L.; Christensen, L.; Duffy, M. J. (1997) The Urokinase-Type Plasminogen Activator System in Cancer Metastasis: A Review. Int. J. Cancer 72, 1-22.
View at Publisher | View at Google Scholar - Hajduk, Philip J., Steven Boyd, David Nettesheim, Vicki Nienaber, Jean Severin, Richard Smith, Don Davidson, Todd Rockway, and Stephen W. Fesik. (2000)
View at Publisher | View at Google Scholar - Andreasen, P. A.; Kjøller, L.; Christensen, L.; Duffy, M. J. (1997) The Urokinase-Type Plasminogen Activator System in Cancer Metastasis: A Review. Int. J. Cancer 72, 1-22.
View at Publisher | View at Google Scholar - Schmitt, M.; Goretzki, L.; Jänicke, F.; Calvete, J.; Eulitz, M.; Kobayashi, H.; Chucholowski, N.; Graeff, H. (1991) Biological and Clinical Relevance of the Urokinase-Type Plasminogen Activator (uPA) in Breast Cancer. Biomed. Biochim. Acta 50, 731-741.
View at Publisher | View at Google Scholar - Spraggon, G.; Phillips, C.; Nowak, U. K.; Ponting, C. P.; Saunders, D.; Dobson, C. M.; Stuart, D. I.; Jones, E. Y. (1995) The Crystal Structure of the Catalytic Domain of Human Urokinase-Type Plasminogen Activator. Structure 3, 681-691.
View at Publisher | View at Google Scholar - Plesner, T.; Behrendt, N.; Ploug, M. (1997) Structure, Function and Expression on Blood and Bone Marrow Cells of the Urokinase-Type Plasminogen Activator Receptor, uPAR. Stem Cells 15, 398- 408.
View at Publisher | View at Google Scholar - Towle, M. J.; Lee, A.; Maduakor, E. C.; Schwartz, C. E.; Bridges, A. J.; Littlefield, B. A. Cancer Res. (1993), 53, 2553. 12- Subasinghe, Nalin L., Carl Illig, James Hoffman, M. Jonathan Rudolph, Kenneth J. Wilson, Richard Soll, Troy
View at Publisher | View at Google Scholar - Randle et al. (2001)
View at Publisher | View at Google Scholar - Geyer, A. G.; Mcclellan, W. J.; Rockway, T. W.; Stewart, K. D.; Weitzberg, M.; Wendt, M. D. (1999) International Patent Application WO 99/05096; Chem. Abstr. 130, 153476.
View at Publisher | View at Google Scholar - Barber, C. G.; Fish, P. V.; Dickinson, R. P. (1999) International Patent Application WO 99/20608; Chem. Abstr. 130, 311705.
View at Publisher | View at Google Scholar - S. Ulisse, E. Baldini, S. Sorrenti, and M. D’Armiento, (2009) “The urokinase plasminogen activator system: a target for anti-cancer therapy,” Current Cancer Drug Targets, vol. 9, no. 1, pp. 32–71.
View at Publisher | View at Google Scholar - G. Spraggon, C. Phillips, U. K. Nowak et al., (1995) “The crystal structure of the catalytic domain of human urokinase- type plasminogen activator,” Structure, vol. 3, no. 7, pp. 681–691,
View at Publisher | View at Google Scholar - Sulimov, V. B., E. V. Katkova, I. V. Oferkin, A. V. Sulimov, A. N. Romanov, A. I. Roschin, I. B. Beloglazova, O. S. Plekhanova, V. A. Tkachuk, and V. A. Sadovnichiy. (2014)
View at Publisher | View at Google Scholar - Wendt, Michael D., Todd W. Rockway, Andrew Geyer, William McClellan, Moshe Weitzberg, Xumiao Zhao, Robert Mantei et al. (2004)
View at Publisher | View at Google Scholar - Nienaber, Vicki L., Donald Davidson, Rohinton Edalji, Vincent L. Giranda, Vered Klinghofer, Jack Henkin, Peter Magdalinos et al. (2000)
View at Publisher | View at Google Scholar - Allwood, Matthew B., Booma Cannan, Daan MF van Aalten, and Ian M. Eggleston. (2007):
View at Publisher | View at Google Scholar - Spargon, G.
View at Publisher | View at Google Scholar - Zajac, Matthew A., Anthony G. Zakrzewski, Mark G. Kowal, and Saraswathi Narayan. (2003)
View at Publisher | View at Google Scholar - Bajaber, Majed Abdullah. (2014).
View at Publisher | View at Google Scholar - Miyamoto, Yu, Yuki Yamada, Hayato Shimazaki, Kazuaki Shimada, Toshiki Nokami, Keiji Nishiwaki, Shigenori KASHIMURA, and Kouichi Matsumoto.
View at Publisher | View at Google Scholar
