

Research Article | DOI: https://doi.org/10.31579/2835-835X/022
Antiviral Agents in Hepatitis B virus Systematic Review Structural Analysis of Drug Resistance Mechanisms
1 Head Regulatory Scientific researchIPL research centre Luckow, India
2 Amity University Lucknow India
*Corresponding Author: Krishnasarmapathy, Diagnosis and Treatment Center, Dr Victor Babes”, Bucharest, Romania.
Citation: Krishnasarmapathy, Saihiti Sarma Pathy, (2023), Antiviral Agents in Hepatitis B Virus Systematic Review Structural Analysis of Drug Resistance Mechanisms, Clinical Trials and Case Studies, 2(2); DOI:10.31579/2835-835X/022
Copyright: © 2023, Krishnasarmapathy. This is an open-access artic le distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 05 April 2023 | Accepted: 15 April 2023 | Published: 20 April 2023
Keywords: hepatitis B virus; anti-HBV agents; drug resistance
Abstract
Tenofovir (TFV) is a widely used treatment for chronic hepatitis B virus (HBV) infection. There is a high genetic barrier to the selection of TFV resistance-associated mutations (RAMs), but the distribution and clinical significance of TFV RAMs are not well understood We carried out a systematic literature search. since the first FDA approval of Lamivudine in 1998, many nucleo(t)side analogs such as Lamivudine, Adefovir, and Entecavir have been used. However, they only inhibit DNA synthesis, and if their administration is stopped a viral breakthrough can develop, making long-term administration necessary, ultimately followed by the development of resistance. Tenofovir has been developed and drug-resistant mutations have decreased significantly, but the problem of resistance due to long-term drug use still remains, along with the drug safety problem. In this review, we introduce the recent trend in the development of hepatitis B treatment agents treatment for hepatitis B (drug repositioning) without resistance and which target the various life cycles of HBV.
Introduction
Over 300 million people, about 4% of the world’s population, are chronically infected with hepatitis B virus (HBV), and a significant number of these patients also suffer from liver diseases such as cirrhosis and liver cancer. In East Asia (especially Taiwan, Japan, Korea and China), HBV is prevalent, with the numberof infected people in Korea estimated to be over 2 million,accounting for 60~70% of chronic liver diseases (1, 2). In over 60% of the cases in Korea the infection route for chronic hepatitis B is vertical transmission from mother to infant at childbirth. Treatments for hepatitis B are interferon alpha injections including Peg-interferon and orally-administered nucleo(t)side analogs. Interferon agents have a fixed administration period and are expected to improve the biochemical and histological findingsdue to their virus-suppressing effect and immune-modulating action, but their use has declined due to the low treatment reaction and adverse side effects. As for nucleo(t)side analogs, they only inhibit DNA synthesis, and if their administration is stopped a viral breakthrough can develop, making long-term administration necessary, ultimately followed by the develop- ment of resistance. Recently, Tenofovir (TDF) has been developed and drug-resistant mutations have decreased significantly, but the problemof resistance by long-term drug use still remains, along with the drug safety problem, due to the treatment characteristics of chronic hepatitis B (3). As such, this paper examinesthe recent trend in the development of hepatitis B treatment agents. The preparation of Tenofovir disoproxil fumarate is (R)-(1-(6-amino-7H-puri-7-yl) propan-2-yloxy)methyl phosphoric acid undergoes condensation with Chloro methyl isopropyl carbonate in the presence of Nmethyl pyyrolidine as a solvent to give (R)-(((1-(6-amino 9H-purin-9-yl)propan2 yloxy)methyl)phosphoryl)bis(oxy)bis(methylene) isopropyl dicarbonate followed by saltification reaction with fumaric acid in IPA as a solvent media to give Tenofovir disoproxil fumarate
Figure 1: Nucleo(t)side analogues for inhibition of HBV replication.
Characteristics and life cycle of HBV
HBV is a 3.2kbp partially double-strand DNA virus which exists as a complete form of Dane particles with infectivity surrounded by capsid and envelop. The DNA is enclosed by a capsid made of core proteins, which are in turn enclosed by surface proteins. HBV characteristically infects only liver cells (hepatotropic) and causes persistent infection without the degeneration of infected cells (non-cytopathic)1. When the HBV virion infects the liver cells, surface proteins are released. Then the viral gene inside the capsid enters into the nucleus and the partially double-stranded HBV DNA transforms into a circulartype of cccDNA. The viral RNA produced from the cccDNAproduces core and surface proteinsand polymerase, and encapsidation progresses in the cytoplasm with pre-genomic RNA (pgRNA), which can be converted to the original viral DNA. After the conversion from pgRNA to DNA, the HBV virion goes through the budding process, after which it can infect or re-infectthe surrounding liver cells, causingpersistent proliferation (4). pgRNA is transcribed to the DNA within the capsid, and at this time nucleoside analogs cut in to the newly synthesized DNA strand anterminate the synthesis. HBV polymerase consists of four domains, and various drugs have been developed to target the reverse transcriptase (RT) domain where DNA is synthesized from RNA [2-4] and the structural changes of the active site due to mutations of the RT domain are the cause of drug resistance (Fig. 1).
Discussion
Summary of key findings
TFV is a safe and effectivetreatment choice for CHB in the majority of cases, and large case series have not raised significant concerns about clinically significant drug resistance. However,it is important to considerthe potential for the emergenceof
resistance, demonstrated by persistent viraemiaon therapy and/or reduced virologic suppression in vitro. Based on existing evidence, TFV resistance seems likely to depend on the selec-tion of suites of mutations (most commonly including L180M, A181V/T, M204I/V and/or N236T), overlapping with RAMs that allow escapefrom other NA drugs. Thereis also a suggestion that,rarely, single mutationscan confer TFV resistance, best demonstrated for S78T [5-6].
Notably, the literature to date is limited and heterogenous, and there remainsa lack of evidence about the frequencyand likely impact of proposed TFV RAMs, either within individual patients or at population level. At present,we have tackledthis uncertainty by dividing our catalogue of polymorphisms into a ‘long-list’ (all reported RAMs) and a ‘short-list’ (RAMs with the best evidence-base of support).
Tools that have been designed to identify drug resistance may bias againstdetection of relevantmutations if they do not scrutinise all relevant sites that contribute to reducing TFV susceptibility. For example, ‘TRUGENE’a commercially availableHBV drug resistance interpretation system, captures
Figure 2: Cartoon to show the sites of TFV drug resistance polymorphisms, using the homologous crystal structure of HIV RT as a model.
The sequence alignment of HBV was extended with HIV RT’s p66 domain and then projected onto a high-resolution HIV RT structure (PDB code 3dlk). Sub-domains of the HIV RT are coloured and annotated. Positions associated with resistance are scattered primarily throughout the finger and palm subdomains of the p66 domain (purplespace-filled representations, left whole-molecule view,purple stick representation on the zoomed in view on the right). Three aspartate residues, D83, D205 and D206 (indicated by grey space- filled representation) form the catalytic triad of the enzyme and are shown as a point of reference. Of the 37 sites identified as potential TFV RAMs, 24 residues which are visible in the structure are labelled (using HBV numbering). This excludes seven putative HBV mutations atsites which do not have a homologous site in the HIV structure (sites 78, 80, 130, 134, 153, 163 and 256), and six sites which are beyond the end of the sequence of the solved crystal HIV structure (267, 269, 278, 317, 333 and 337). Figure producedusing the ICM platform. Note that in most cases, individual mutationsare unlikely to be sufficient to mediate resistance, and a resistantphenotype arises only as a result of combinations of 2 polymorphisms. HBV, hepatitis B virus; HIV, human immunodeficiency virus; RAM, resistance-associated mutation;RT, reverse transcriptase; TFV, tenofovir. common HBV RAMs but does not include positions 78, 177, or 249 which may be pertinent to TFV resistance [39] and ‘geno2pheno hbv’ only lists one TFV mutation at position 236.
Overlap of TFV RAMs with RAMs to other NA agents RAMs L180M, M204I/V and A181T/V have been associ- ated with resistance to 3TC, telbivudine (LdT) and entecavir(ETV)3{40-43]. their reportedassociation with TFV resistance is of concernin suggesting that prior NA exposure can increase the likelihood of cross-resistance to TFV. A study of HIV/HBV co-infected individuals demonstrated a decreased likelihoodof HBV DNA suppression with TDF among individuals exposedto prolonged 3TC treatment, possiblydue to accumulation of such mutations [44]. A large study in China reportedA181 and/ or N236 substitutions in 11% of the population [42], which may underpinreduced susceptibility to TFV. The structural similari- ties between ADV and TFV, and similarinteraction with HBV polymerase [1,2] explain why the ADV RAMs A181T/Vand N236T are also reported to confer resistance to TFV [1,42].
Although TFV has been considered effective in the contextof resistance to other NAs [36], the current evidencesuggests that there may be common pathwaysto resistance [37]. Thereis some evidence showing co-location of RAMs conferring resistance to differentantiviral agents on the same viral haplotype [38]. These findings suggesting cross-resistance are of concern, especially for settings in which there has been widespread use of NA therapy as a component of ART for HIV [3].
Sites of TFV RAMs in HBV RT
Resistance to TFV can be explained by RAMs both within and outside the activesite of the RT enzyme,some of which may have similar mechanisms to those describedin HIV [10,38]. The mechanism of resistance in most of these polymorphisms remains unknown, but may interfere with drug access to sites of activitythrough steric hindrance. Mutations within activesites of the enzyme may be associated with a higherfitness cost to the virus than mutations at other locations in the RT sequence, as they are more likelyto interfere with the RT function. Some polymorphisms listed as RAMs may in fact representcompensatory mutations, which are co-selected in the pres- ence of primary RAMs. For example,substitution at position269 has been previously described as a compensatory mutation that restores impairments to RT function [39].
Currently, HBV genotyping is not routinely undertaken in clini-cal practice, so it is difficult to amass data for any potential relationship between resistance and viral genotype.However, there are some clues that genotype may be relevant. For example, C256S has been linked to TFV resistance, but S256 is wild type in genotypeC (Suppl Table4, Extended data5), suggesting that the geneticbarrier to TFV resistance in genotype C might be lower than in other genotypes. However, a study of >1000 individuals in China found no differences in drug resistance rates between genotypeB vs genotype C infection [42] The identification of Y9H as a TFV RAM should be viewed with cautionas H9 is frequently the wildtype residue,irrespective of genotype.
Other factors associated with persistent vireamia In addition to RAMs, there are other explanations for incom- pletesuppression of HBV viraemia on therapy [22,33], including a higher baselineHBV DNA level, positive baselineHBeAg status, historyof 3TC exposure, a lower nadir CD4+ T cell count in the context of HIV coinfection, and high serum HBV RNA levels44,40,41. Given that HBV DNA is inhibited in a dose- dependent manner [2], it is also possible that insufficient drug delivery to the infectedhepatocyte could be the causeof persistent viraemiaeven in the absence of specific RAMs.
Incomplete adherenceto therapy can also contribute to virological breakthrough [42]. Two studies included in our review assessed treatment compliance by measuring drug concentra- tion in plasma [9,31]. Assessment of adherence in chronic HBV has been through the use of questionnaires [43], but these are subject to self-reporting bias. Evidence of potential TFV resist- ance may emerge when individuals with HIV/HBV coinfection are treated with a TFV-containing regimen leading to suppression of HIV but with sustained HBV vireamia [46]
It has been reported to take three years for 90% of HBV infected individuals to reach viraemicsuppression on therapy [55], in contrast to HIV, in which 88% of patientssuppress the viruswithin the first year of TDF-based treatment [56]. In the studies we have reportedin this review, persistent HBV viraemia on therapy could be due to the prolonged timelinefor viraemic suppression; however, in most studies there was a reduction in viral load when TDF was initiated, with subsequent virological breakthrough that is more in keeping with the selectionof resistance.
Caveatsand limitations
There is sparse literature on HBV resistance to TFV, and stud- ies are of varying quality. While there is a high genetic barrier to selection of TFV resistance, it is likely that there is under-reporting of cases of resistance, particularly in low/middle incomeset- tings in which routine monitoring of HBV viral load on treatment is not undertaken. It can be difficult to infer the impact of common polymorphisms on drug resistance phenotype; for example,it is plausible that M204I/Vmay be enriched among TFV resistant strainssimply as a ‘footprint’ of prior exposure to 3TC.
Most studiesto date have used Sanger sequencing, and it is possible that significant minorityvariants may be under- represented, as suggested by one reportin which phenotypic TFV resistance was associated with RAMs in <20>
We recognisethe limitations of drawing direct comparisons between HIV and HBV RT, given the limited (<30>
HBV treatment agents
The full-scale treatment of chronic hepatitis B began in the early 1990s when interferon was first used (5). Then, after the first FDA approval of Lamivudine (LMV) in 1998, nucleo(t)side analogssuch as Adefovir (ADV), Entecavir (ETV), Telbivudine (LdT), Clevudine (CLV), and Tenofovir (TDF) came to be used worldwide (Table 1) (6). Despite the various advantages of interferon (IFN-alpha), it is inconvenient to use as an injection and is of limited use for patients with decompensated cirrhosis; thus, orally administered antiviral agents are currently the mainstream treatment for chronic hepatitis B. As tenofovir alafenamide fumarate (TAF) and besifovir dipivoxil maleate (Besifovir, BSV) were approved as a treatment medication for adults with chronic hepatitisB in South Korea in 2017, a total of eight antiviralagents can now be used (Figure. 2) [7-15].al. 2018)
Table 1: HBV treatmentdrugs available for chronic Hepatitis B virus (HBV)infection (Modified from Tang LSY et
Figure 2: Advances in HBV treatment.
Lamivudine (LMV, 3TC)
LMV was originally developed for AIDS treatment as an inhibitorfor HIV reversetranscriptase, and was also approvedas a hepatitis B treatment drug, i.e. a 1st-generation nucleotide analogue, by the FDA in 1998 because it effectively inhibits HBV reverse transcriptase (7). LMV is successively phosphorylated to LMV triphosphate by intracellular kinase,and shortly after the diphosphate groups are eliminated, LMV 5’-monophosphate is incorporated into the newly produced viral DNA by HBV polymerase. Because it is a nucleoside analog without a 3’-OH group for chain polymerization, it induces the termination of polymerization synthesis and inhibits viral replication [16]. Although the action mechanism of most antiviral agents is similar to this, drug-resistant mutation within five years is as high as 70%.
Adefovir (ADV)
ADV, an adenosinemonophosphate analogue, is phosphorylated by intracellular kinaseand activated to ADV diphosphate, whereupon it competitively inhibits the reaction deoxyadenosine triphosphate, a substrate of HBV DNA polymerase. ADV diphosphate is incorporated into newly producedviral DNA, and has an antiviral effect caused by a similaraction mechanism to that of LMV, but the recommended dosage is 1/10 (10 mg/day) that of LMV, and its rate of drug-resistant mutation within5 years has been improvedby 20-29% (6). Although ADV had an important role for LMV-resistant patients, it is no longerrecommended as a first-line therapy [17-18].
Entecavir (ETV)
ETV, a guanosine nucleoside analogue approvedby the US FDA in March 2005, is known to show the more enhanced drug effect (0.5~1 mg/day) compared to other existing drugs, is less likely to cause an adverse reaction, and has only a 1.2% incidence of drug-resistant mutationwithin five years(8). Because of this excellent resistance rate, ETV is recommended as a first-line therapy for chronicHBV (9).
Telbivudine (LdT)
LdT, a thymidine nucleoside analogue, is an unmodified L-isomer of thymidine, a naturally occurring nucleoside. Thus, the phosphorylation reaction to the active form LdT triphosphate occurs easily. However, LdT has not been used for a first-line therapy [18,19] because of the higher frequencyof resistance in early time during administration similar to LMV [9].
Clevudine (CLV)
CLV approved in 2006 in South Korea, a pyrimidine analogue (30 mg/day), is known to not only inhibit DNA-dependent DNA activity for HBV polymerase but also to demonstrate an antiviral effect by interrupting reverse transcription and priming.CLV is about 10-fold more potent than LMV againstHBV19 in cell culture, and HBV DNA level was reduced by 2.5 to 3 log10 copies/mL for 4 weeks trial with 10 to 200 mg/day (10). CLV is being marketed in Korea and Philippines named as Lenovir and Revovir, respectively.
Tenofovir (TDF)
Tenofovir disoproxil fumarate, a prodrug of Tenofovir (TDF),is an oral antiviral agent approved for HIV and HBV treatments (11). TDF is successively phosphorylated by intracellular kinaseand activated to tenofovir diphosphate, and then competeswith endogenous nucleotide deoxyadenosine 5'-triphosphate (dATP) for incorporation into the newly replicated HBV DNA by HBV polymerase. Incorporated TDF, instead of endogenous nucleotide, is a nucleotide analogue without a 3’-OH group,which is required for the elongation of DNA base chains, and thus it induces the termination of polymerization synthesis and inhibits viral replication (12) [20,21] This action mechanism is very similar to that of another nucleotide analogue, ADV, but the antiviral efficacy of TDF is much stronger than that of ADV because the latter is used in a limited dosage (only 10 mg) so as to reduce the development of nephrotoxicity, whereas TDF is used in a far higher dose of 300 mg (13). Also, it is assumed that the higher binding affinity of TDF to HBV polymerase, compared to ADV, is related to its strong efficacy (14). Drug-resistant mutationwas not reportedinitially, but mutationshave been observedmore recently (6, 15).
Besifovir (LB80380)
Besifovir is an acyclic nucleotide phosphonate similar to Adefovir or Tenofovir. It is a nucleotide analogue and a prodrug of guanosine triphosphate nucleotide analogue LB80317. Besifovir is absorbed into the intestine and deacetylated to the intermediate metabolite LB80331 by esterase in the intestine and the liver, and then oxidized to the active metabolite LB80317 by oxidase (aldehyde oxidase or xanthine oxidase). In the liver cells it is phosphorylated to diphosphate and triphosphate forms, after which it competes with dGTP to bind with HBV DNA polymerase, and thus blocks the action of polymerase and inhibits viral proliferation. After the phase 2 clinical trial conducted. It was released on the marketin 2017 as a domesticnovel drug, witha similar efficacyto that of TDF, but without the latter’s adverseside effect of reduced bone density (16) [22].
Tenofovir alafenamide fumarate (TAF, GS7340)
TAF, a prodrug of Tenofovir used in the body and a nucleotide analogue like TDF, is an oral antiviral agent that inhibits reverse transcription from pre-genomic RNA to DNA. Prodrugs are being developed to reduce its adverse effects on the functions of the kidneys by increasing bioactivity and enhancing the antiviral action compared to the strong antiviral agent TDF. The most representative drug is TAF. TAF is converted to tenofovir-alanine conjugate (TAF-Ala) in the body and then converted again to TDF and phosphorylated to active metabolite tenofovir diphosphate (TAF-DP)for drug action.Recently, the results of a phase 3 clinical trials for comparison with TDF in HIV patients showed similar antiviral effects, but TAF showed significantly better responses compared with the disadvantages of TDF, such as increased serum creatine, proteinuria, and reduced bone density. In addition, studies on CMX157, a hexadecyloxypropyl conjugate of TDF, and on AGX-1009 [23] of another structure, are currently in progress.
HBV drug resistance
For the effective treatment of viruses, drugs are administered by monotherapy or combination therapy. Methods with the least incidenceof resistant virusesand a quick treatment for HBV have been widelystudied and are currently being applied in clinical situations.
The long-term administration (i.e. more than one year) of a medication for chronic HBV infection [24] leads to the development of drug-resistant mutation viruses in many cases. This is because there is an active site in the center of HBV polymerase where DNA synthesis occurs; if mutations of the amino acids inherent in each drug occur near this site, drugs cannot incorporate into the site due to steric hindrance, whereupon resistance develops. Ultimately, it is most important to select the optimal antiviral agent for treatment by continuously monitoring each drug for the development of a mutation after its administration, because each drug displays different resistance patterns and mutations during long-term administration. The characteristics of resistance for each antiviralagent studied up to the present time are as follows (Fig. 3).
Figure 3: HBVresistance to nucleo(t)side analogues.
Lamivudine resistance
The incidence of resistant viruses during LMV treatment has increased by 14~32
Discussion
Summary of key findings
TFV is a safe and effectivetreatment choice for CHB in the majority of cases, and large case series have not raised significant concerns about clinically significant drug resistance. However,it is important to considerthe potential for the emergenceof
resistance, demonstrated by persistent viraemiaon therapy and/or reduced virologic suppression in vitro. Based on existing evidence, TFV resistance seems likely to depend on the selec-tion of suites of mutations (most commonly including L180M, A181V/T, M204I/V and/or N236T), overlapping with RAMs that allow escapefrom other NA drugs. Thereis also a suggestion that,rarely, single mutationscan confer TFV resistance, best demonstrated for S78T [5-6].
Notably, the literature to date is limited and heterogenous, and there remainsa lack of evidence about the frequencyand likely impact of proposed TFV RAMs, either within individual patients or at population level. At present,we have tackledthis uncertainty by dividing our catalogue of polymorphisms into a ‘long-list’ (all reported RAMs) and a ‘short-list’ (RAMs with the best evidence-base of support).
Tools that have been designed to identify drug resistance may bias againstdetection of relevantmutations if they do not scrutinise all relevant sites that contribute to reducing TFV susceptibility. For example, ‘TRUGENE’a commercially availableHBV drug resistance interpretation system, captures
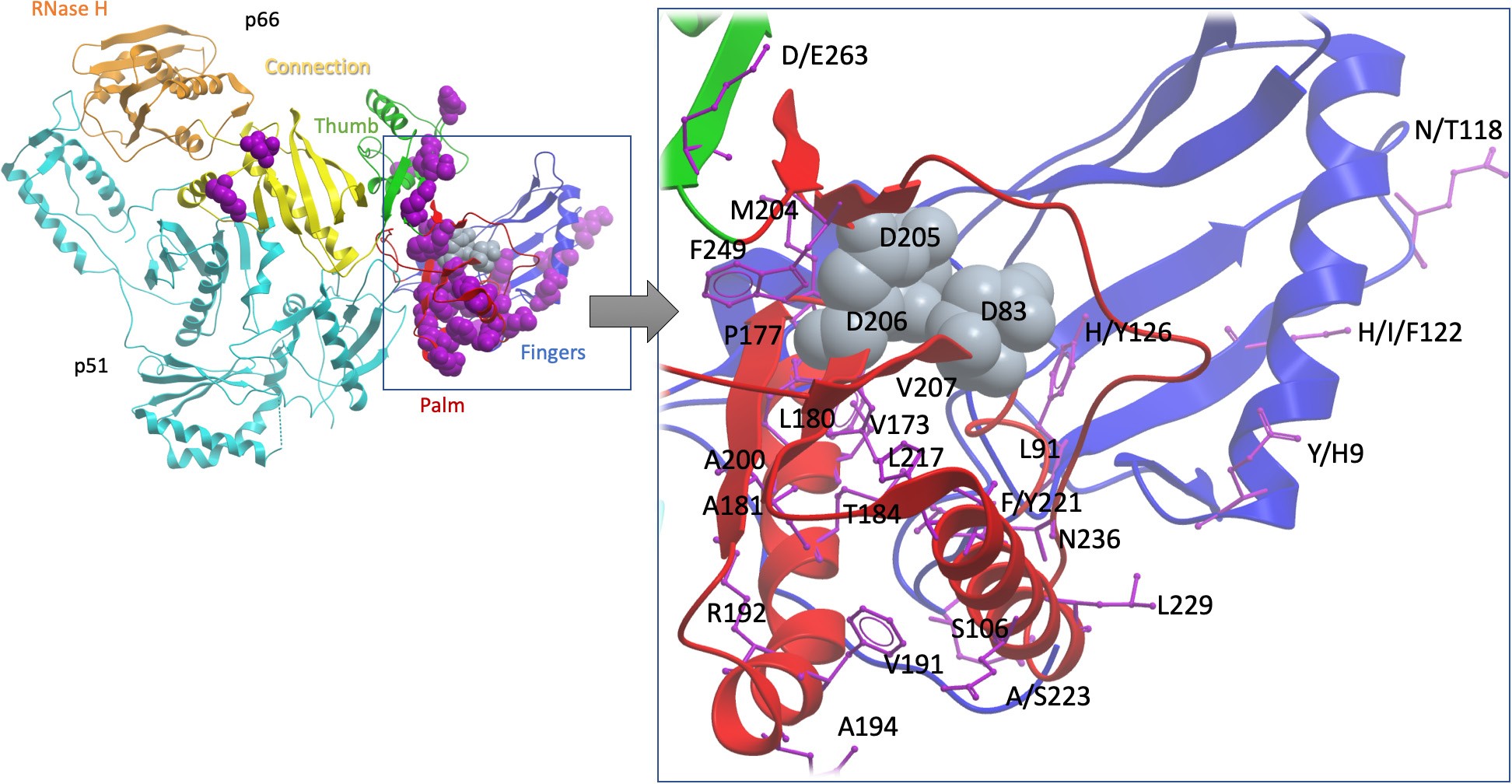
Figure 2: Cartoon to show the sites of TFV drug resistance polymorphisms, using the homologous crystal structure of HIV RT as a model.
The sequence alignment of HBV was extended with HIV RT’s p66 domain and then projected onto a high-resolution HIV RT structure (PDB code 3dlk). Sub-domains of the HIV RT are coloured and annotated. Positions associated with resistance are scattered primarily throughout the finger and palm subdomains of the p66 domain (purplespace-filled representations, left whole-molecule view,purple stick representation on the zoomed in view on the right). Three aspartate residues, D83, D205 and D206 (indicated by grey space- filled representation) form the catalytic triad of the enzyme and are shown as a point of reference. Of the 37 sites identified as potential TFV RAMs, 24 residues which are visible in the structure are labelled (using HBV numbering). This excludes seven putative HBV mutations atsites which do not have a homologous site in the HIV structure (sites 78, 80, 130, 134, 153, 163 and 256), and six sites which are beyond the end of the sequence of the solved crystal HIV structure (267, 269, 278, 317, 333 and 337). Figure producedusing the ICM platform. Note that in most cases, individual mutationsare unlikely to be sufficient to mediate resistance, and a resistantphenotype arises only as a result of combinations of 2 polymorphisms. HBV, hepatitis B virus; HIV, human immunodeficiency virus; RAM, resistance-associated mutation;RT, reverse transcriptase; TFV, tenofovir. common HBV RAMs but does not include positions 78, 177, or 249 which may be pertinent to TFV resistance [39] and ‘geno2pheno hbv’ only lists one TFV mutation at position 236.
Overlap of TFV RAMs with RAMs to other NA agents RAMs L180M, M204I/V and A181T/V have been associ- ated with resistance to 3TC, telbivudine (LdT) and entecavir(ETV)3{40-43]. their reportedassociation with TFV resistance is of concernin suggesting that prior NA exposure can increase the likelihood of cross-resistance to TFV. A study of HIV/HBV co-infected individuals demonstrated a decreased likelihoodof HBV DNA suppression with TDF among individuals exposedto prolonged 3TC treatment, possiblydue to accumulation of such mutations [44]. A large study in China reportedA181 and/ or N236 substitutions in 11% of the population [42], which may underpinreduced susceptibility to TFV. The structural similari- ties between ADV and TFV, and similarinteraction with HBV polymerase [1,2] explain why the ADV RAMs A181T/Vand N236T are also reported to confer resistance to TFV [1,42].
Although TFV has been considered effective in the contextof resistance to other NAs [36], the current evidencesuggests that there may be common pathwaysto resistance [37]. Thereis some evidence showing co-location of RAMs conferring resistance to differentantiviral agents on the same viral haplotype [38]. These findings suggesting cross-resistance are of concern, especially for settings in which there has been widespread use of NA therapy as a component of ART for HIV [3].
Sites of TFV RAMs in HBV RT
Resistance to TFV can be explained by RAMs both within and outside the activesite of the RT enzyme,some of which may have similar mechanisms to those describedin HIV [10,38]. The mechanism of resistance in most of these polymorphisms remains unknown, but may interfere with drug access to sites of activitythrough steric hindrance. Mutations within activesites of the enzyme may be associated with a higherfitness cost to the virus than mutations at other locations in the RT sequence, as they are more likelyto interfere with the RT function. Some polymorphisms listed as RAMs may in fact representcompensatory mutations, which are co-selected in the pres- ence of primary RAMs. For example,substitution at position269 has been previously described as a compensatory mutation that restores impairments to RT function [39].
Currently, HBV genotyping is not routinely undertaken in clini-cal practice, so it is difficult to amass data for any potential relationship between resistance and viral genotype.However, there are some clues that genotype may be relevant. For example, C256S has been linked to TFV resistance, but S256 is wild type in genotypeC (Suppl Table4, Extended data5), suggesting that the geneticbarrier to TFV resistance in genotype C might be lower than in other genotypes. However, a study of >1000 individuals in China found no differences in drug resistance rates between genotypeB vs genotype C infection [42] The identification of Y9H as a TFV RAM should be viewed with cautionas H9 is frequently the wildtype residue,irrespective of genotype.
Other factors associated with persistent vireamia In addition to RAMs, there are other explanations for incom- pletesuppression of HBV viraemia on therapy [22,33], including a higher baselineHBV DNA level, positive baselineHBeAg status, historyof 3TC exposure, a lower nadir CD4+ T cell count in the context of HIV coinfection, and high serum HBV RNA levels44,40,41. Given that HBV DNA is inhibited in a dose- dependent manner [2], it is also possible that insufficient drug delivery to the infectedhepatocyte could be the causeof persistent viraemiaeven in the absence of specific RAMs.
Incomplete adherenceto therapy can also contribute to virological breakthrough [42]. Two studies included in our review assessed treatment compliance by measuring drug concentra- tion in plasma [9,31]. Assessment of adherence in chronic HBV has been through the use of questionnaires [43], but these are subject to self-reporting bias. Evidence of potential TFV resist- ance may emerge when individuals with HIV/HBV coinfection are treated with a TFV-containing regimen leading to suppression of HIV but with sustained HBV vireamia [46]
It has been reported to take three years for 90% of HBV infected individuals to reach viraemicsuppression on therapy [55], in contrast to HIV, in which 88% of patientssuppress the viruswithin the first year of TDF-based treatment [56]. In the studies we have reportedin this review, persistent HBV viraemia on therapy could be due to the prolonged timelinefor viraemic suppression; however, in most studies there was a reduction in viral load when TDF was initiated, with subsequent virological breakthrough that is more in keeping with the selectionof resistance.
Caveats and limitations
There is sparse literature on HBV resistance to TFV, and stud- ies are of varying quality. While there is a high genetic barrier to selection of TFV resistance, it is likely that there is under-reporting of cases of resistance, particularly in low/middle incomeset- tings in which routine monitoring of HBV viral load on treatment is not undertaken. It can be difficult to infer the impact of common polymorphisms on drug resistance phenotype; for example,it is plausible that M204I/Vmay be enriched among TFV resistant strainssimply as a ‘footprint’ of prior exposure to 3TC.
Most studiesto date have used Sanger sequencing, and it is possible that significant minorityvariants may be under- represented, as suggested by one reportin which phenotypic TFV resistance was associated with RAMs in <20>
We recognisethe limitations of drawing direct comparisons between HIV and HBV RT, given the limited (<30>
HBV treatment agents
The full-scale treatment of chronic hepatitis B began in the early 1990s when interferon was first used (5). Then, after the first FDA approval of Lamivudine (LMV) in 1998, nucleo(t)side analogssuch as Adefovir (ADV), Entecavir (ETV), Telbivudine (LdT), Clevudine (CLV), and Tenofovir (TDF) came to be used worldwide (Table 1) (6). Despite the various advantages of interferon (IFN-alpha), it is inconvenient to use as an injection and is of limited use for patients with decompensated cirrhosis; thus, orally administered antiviral agents are currently the mainstream treatment for chronic hepatitis B. As tenofovir alafenamide fumarate (TAF) and besifovir dipivoxil maleate (Besifovir, BSV) were approved as a treatment medication for adults with chronic hepatitisB in South Korea in 2017, a total of eight antiviralagents can now be used (Figure. 2) [7-15].al. 2018)

Table 1: HBV treatmentdrugs available for chronic Hepatitis B virus (HBV)infection (Modified from Tang LSY et
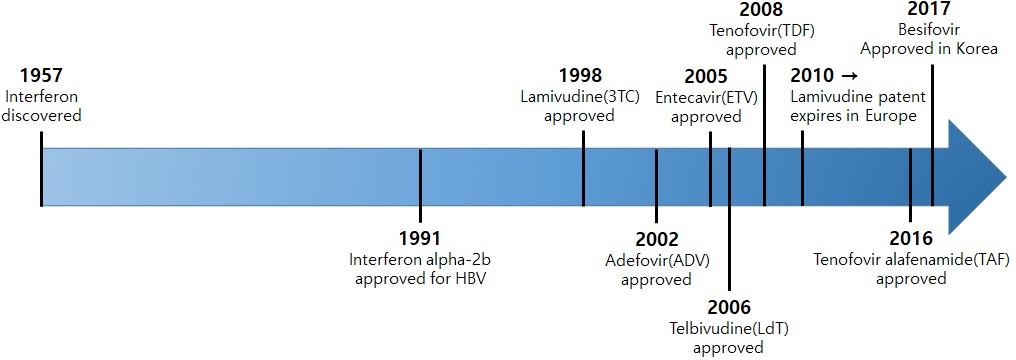
Figure 2: Advances in HBV treatment.
Lamivudine (LMV, 3TC)
LMV was originally developed for AIDS treatment as an inhibitorfor HIV reversetranscriptase, and was also approvedas a hepatitis B treatment drug, i.e. a 1st-generation nucleotide analogue, by the FDA in 1998 because it effectively inhibits HBV reverse transcriptase (7). LMV is successively phosphorylated to LMV triphosphate by intracellular kinase,and shortly after the diphosphate groups are eliminated, LMV 5’-monophosphate is incorporated into the newly produced viral DNA by HBV polymerase. Because it is a nucleoside analog without a 3’-OH group for chain polymerization, it induces the termination of polymerization synthesis and inhibits viral replication [16]. Although the action mechanism of most antiviral agents is similar to this, drug-resistant mutation within five years is as high as 70%.
Adefovir (ADV)
ADV, an adenosinemonophosphate analogue, is phosphorylated by intracellular kinaseand activated to ADV diphosphate, whereupon it competitively inhibits the reaction deoxyadenosine triphosphate, a substrate of HBV DNA polymerase. ADV diphosphate is incorporated into newly producedviral DNA, and has an antiviral effect caused by a similaraction mechanism to that of LMV, but the recommended dosage is 1/10 (10 mg/day) that of LMV, and its rate of drug-resistant mutation within5 years has been improvedby 20-29% (6). Although ADV had an important role for LMV-resistant patients, it is no longerrecommended as a first-line therapy [17-18].
Entecavir (ETV)
ETV, a guanosine nucleoside analogue approvedby the US FDA in March 2005, is known to show the more enhanced drug effect (0.5~1 mg/day) compared to other existing drugs, is less likely to cause an adverse reaction, and has only a 1.2% incidence of drug-resistant mutationwithin five years(8). Because of this excellent resistance rate, ETV is recommended as a first-line therapy for chronicHBV (9).
Telbivudine (LdT)
LdT, a thymidine nucleoside analogue, is an unmodified L-isomer of thymidine, a naturally occurring nucleoside. Thus, the phosphorylation reaction to the active form LdT triphosphate occurs easily. However, LdT has not been used for a first-line therapy [18,19] because of the higher frequencyof resistance in early time during administration similar to LMV [9].
Clevudine (CLV)
CLV approved in 2006 in South Korea, a pyrimidine analogue (30 mg/day), is known to not only inhibit DNA-dependent DNA activity for HBV polymerase but also to demonstrate an antiviral effect by interrupting reverse transcription and priming.CLV is about 10-fold more potent than LMV againstHBV19 in cell culture, and HBV DNA level was reduced by 2.5 to 3 log10 copies/mL for 4 weeks trial with 10 to 200 mg/day (10). CLV is being marketed in Korea and Philippines named as Lenovir and Revovir, respectively.
Tenofovir (TDF)
Tenofovir disoproxil fumarate, a prodrug of Tenofovir (TDF),is an oral antiviral agent approved for HIV and HBV treatments (11). TDF is successively phosphorylated by intracellular kinaseand activated to tenofovir diphosphate, and then competeswith endogenous nucleotide deoxyadenosine 5'-triphosphate (dATP) for incorporation into the newly replicated HBV DNA by HBV polymerase. Incorporated TDF, instead of endogenous nucleotide, is a nucleotide analogue without a 3’-OH group,which is required for the elongation of DNA base chains, and thus it induces the termination of polymerization synthesis and inhibits viral replication (12) [20,21] This action mechanism is very similar to that of another nucleotide analogue, ADV, but the antiviral efficacy of TDF is much stronger than that of ADV because the latter is used in a limited dosage (only 10 mg) so as to reduce the development of nephrotoxicity, whereas TDF is used in a far higher dose of 300 mg (13). Also, it is assumed that the higher binding affinity of TDF to HBV polymerase, compared to ADV, is related to its strong efficacy (14). Drug-resistant mutationwas not reportedinitially, but mutationshave been observedmore recently (6, 15).
Besifovir (LB80380)
Besifovir is an acyclic nucleotide phosphonate similar to Adefovir or Tenofovir. It is a nucleotide analogue and a prodrug of guanosine triphosphate nucleotide analogue LB80317. Besifovir is absorbed into the intestine and deacetylated to the intermediate metabolite LB80331 by esterase in the intestine and the liver, and then oxidized to the active metabolite LB80317 by oxidase (aldehyde oxidase or xanthine oxidase). In the liver cells it is phosphorylated to diphosphate and triphosphate forms, after which it competes with dGTP to bind with HBV DNA polymerase, and thus blocks the action of polymerase and inhibits viral proliferation. After the phase 2 clinical trial conducted. It was released on the marketin 2017 as a domesticnovel drug, witha similar efficacyto that of TDF, but without the latter’s adverseside effect of reduced bone density (16) [22].
Tenofovir alafenamide fumarate (TAF, GS7340)
TAF, a prodrug of Tenofovir used in the body and a nucleotide analogue like TDF, is an oral antiviral agent that inhibits reverse transcription from pre-genomic RNA to DNA. Prodrugs are being developed to reduce its adverse effects on the functions of the kidneys by increasing bioactivity and enhancing the antiviral action compared to the strong antiviral agent TDF. The most representative drug is TAF. TAF is converted to tenofovir-alanine conjugate (TAF-Ala) in the body and then converted again to TDF and phosphorylated to active metabolite tenofovir diphosphate (TAF-DP)for drug action.Recently, the results of a phase 3 clinical trials for comparison with TDF in HIV patients showed similar antiviral effects, but TAF showed significantly better responses compared with the disadvantages of TDF, such as increased serum creatine, proteinuria, and reduced bone density. In addition, studies on CMX157, a hexadecyloxypropyl conjugate of TDF, and on AGX-1009 [23] of another structure, are currently in progress.
HBV drug resistance
For the effective treatment of viruses, drugs are administered by monotherapy or combination therapy. Methods with the least incidenceof resistant virusesand a quick treatment for HBV have been widelystudied and are currently being applied in clinical situations.
The long-term administration (i.e. more than one year) of a medication for chronic HBV infection [24] leads to the development of drug-resistant mutation viruses in many cases. This is because there is an active site in the center of HBV polymerase where DNA synthesis occurs; if mutations of the amino acids inherent in each drug occur near this site, drugs cannot incorporate into the site due to steric hindrance, whereupon resistance develops. Ultimately, it is most important to select the optimal antiviral agent for treatment by continuously monitoring each drug for the development of a mutation after its administration, because each drug displays different resistance patterns and mutations during long-term administration. The characteristics of resistance for each antiviralagent studied up to the present time are as follows (Fig. 3).
 |
Figure 3: HBVresistance to nucleo(t)side analogues.
Lamivudine resistance
The incidence of resistant viruses during LMV treatment has increased by 14~32
Conclusion
There is emergingevidence for polymorphisms that may reduce susceptibility to TVF. However,good correlation between viral sequenceand treatment outcomesis currently lacking;further studies are essential to optimise individual treatment and public health approaches. Currently used antiviral drugs for HBV are viral polymerase inhibitors. LMV has mainly been used as an initial treatment drug for chronic hepatitis B, while ADV has been used as an alternative for LMV resistance. As shown in Fig. 3, the long-term use of HBV treatment drugs can induce drug-resistant mutation within the active site of the reverse transcriptase domainof HBV. Compared to the antiviral agentssuch as ADV, ETV, and LdT, TDF has been used becausethese are strongagents with less resistance even during long-term administration, but further studies are necessary whether recentlyreported mutations are clinically significant.Currently available drugs are unable to achieve the complete eradiation of the cccDNA of chronic hepatitis B, resulting to an inactive carrier state with suppressed viral replication and continuous surveillance for liver cancer. Therefore, it is essential to develop antiviral agents with a totally different action mechanism targeting the HBV life cycle, for example, inhibiting the HBV entry to hepatocytes, the HBx interaction, HBV core assembly, and HBV entry to the hepatocytes (29). Also, new immunomodulatory therapies targeting HBV such as Toll-like receptor agonists have been developing for overcoming the host tolerance.
It is no doubt that nucleo(t)side is the first-line therapy, but occurring the mutations during long-term administration are limitation for complete cure of HBV. Further studies on a combination therapy using a nucleo(t)side analogue and novel targeting antiviral agents to HBV life cycle and immune response should be conducted for cure the HBV without the adverse effects.
References
- Kao JH, Chen DS. (2002). Global control of hepatitis B virus infection. Lancet Infect Dis 2:395–403.
View at Publisher | View at Google Scholar - Polaris Observatory Collaborators. (2018). Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol Hepatol 3:383–403.
View at Publisher | View at Google Scholar - Schweitzer A, Horn J, Mikolajczyk RT, Krause G, Ott JJ. (2015). Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet 386:1546–1555.
View at Publisher | View at Google Scholar - Le MH, Yeo YH, Cheung R, Henry L, Lok AS, Nguyen MH. (2019). Chronic hepatitis B prevalence among foreign-born and US-born adults in the United States, 1999-2019. Hepatology
View at Publisher | View at Google Scholar - Mitchell T, Armstrong GL, Hu DJ, Wasley A, Painter JA. (2011). The increasing burden of imported chronic hepatitis B—United States, 1974–2008. PLoS One 6:e27717.
View at Publisher | View at Google Scholar - Nguyen MH, Lim JK, Burak Ozbay A, Fraysse J, Liou I, Meyer N, Dusheiko G, Gordon SC. (2019). Advancing age and comorbidity in a US insured population-based cohort of patients with chronic hepatitis B. Hepatology 69:959–973.
View at Publisher | View at Google Scholar - Liu A, Le A, Zhang J, Wong C, Wong C, Henry L, Nguyen MH. (2018). Increasing co-morbidities in chronic hepatitis B patients: experience in primary care and referral practices during 2000–2015. Clin Transl Gastroenterol 9:141.
View at Publisher | View at Google Scholar - Nguyen MH, Burak Ozbay A, Liou I, Meyer N, Gordon SC, Dusheiko G, Lim JK. (2019). Healthcare resource utilization and costs by disease severity in an insured national sample of US patients with chronic hepatitis B. J Hepatol 70:24–32.
View at Publisher | View at Google Scholar - Lin CL, Kao JH. (2017). Natural history of acute and chronic hepatitis B: the role of HBV genotypes and mutants. Best Pract Res Clin Gastroenterol 31:249–255.
View at Publisher | View at Google Scholar - World Health Organization. (2016). Global health sector strategy on viral hepatitis, 2016-2021, p 56. World Health Organization, Geneva, Switzerland.
View at Publisher | View at Google Scholar - Kao JH. (2015). Hepatitis B vaccination and prevention of hepatocellular carcinoma. Best Pract Res Clin Gastroenterol 29:907–917.
View at Publisher | View at Google Scholar - Chang MH, You SL, Chen CJ, Liu CJ, Lai MW, Wu TC, Wu SF, Lee CM, Yang SS, Chu HC, Wang TE, Chen BW, Chuang WL, Soon MS, Lin CY, Chiou ST, Kuo HS, Chen DS, Taiwan Hepatoma Study Group. 2016. Long-term effects of hepatitis B immunization of infants in preventing liver cancer. Gastroenterology 151:472–480.e1.
View at Publisher | View at Google Scholar - Liaw YF, Sung JJ, Chow WC, Farrell G, Lee CZ, Yuen H, Tanwandee T, Tao QM, Shue K, Keene ON, Dixon JS, Gray DF, Sabbat J, Cirrhosis Asian Lamivudine Multicentre Study Group. (2004). Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med 351:1521–1531.
View at Publisher | View at Google Scholar - en WH, Lai MW, Chang MH. (2016). A review of strategies to prevent mother-to-infant transmission of hepatitis B virus infection. Expert Rev Gastroenterol Hepatol 10:317–330.
View at Publisher | View at Google Scholar - an CQ, Duan Z, Dai E, Zhang S, Han G, Wang Y, Zhang H, Zou H, Zhu B, Zhao W, Jiang H, China Study Group for the Mother-to-Child Transmission of Hepatitis B. 2016. Tenofovir to prevent hepatitis B transmission in mothers with high viral load. N Engl J Med 374:2324–2334. .
View at Publisher | View at Google Scholar - Zou H, Chen Y, Duan Z, Zhang H, Pan C. 2012. Virologic factors associated with failure to passive-active immunoprophylaxis in infants born to HBsAg-positive mothers. J Viral Hepat 19:.
View at Publisher | View at Google Scholar - European Association for the Study of the Liver. 2017. EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J Hepatol 67:370–398.
View at Publisher | View at Google Scholar - Mitchell AE, Colvin HM, Palmer Beasley R. 2010. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology 51:729–733.
View at Publisher | View at Google Scholar - U.S. Department of Health and Human Services. 2011. Combating the silent epidemic of viral hepatitis: action plan for the prevention, care, and treatment of viral hepatitis services. U.S. Department of Health and Human Services, Washington, DC.
View at Publisher | View at Google Scholar - Kallman JB, Arsalla A, Park V, Dhungel S, Bhatia P, Haddad D, Wheeler A, Younossi ZM. 2009. Screening for hepatitis B, C and non-alcoholic fatty liver disease: a survey of community-based physicians. Aliment Pharmacol Ther 29:1019–1024. .
View at Publisher | View at Google Scholar - Foster T, Hon H, Kanwal F, Han S, Spiegel B. 2011. Screening high risk individuals for hepatitis B: physician knowledge, attitudes, and beliefs. Dig Dis Sci 56:3471–3487..
View at Publisher | View at Google Scholar - Ku KC, Li J, Ha NB, Martin M, Nguyen VG, Nguyen MH. 2013. Chronic hepatitis B management based on standard guidelines in community primary care and specialty clinics. Dig Dis Sci 58:3626–3633.
View at Publisher | View at Google Scholar - Nguyen NH, Nguyen V, Trinh HN, Lin B, Nguyen MH. 2013. Treatment eligibility of patients with chronic hepatitis B initially ineligible for therapy. Clin Gastroenterol Hepatol 11:565–571.
View at Publisher | View at Google Scholar - Uribe LA, Nguyen N, Kim L, Trinh HN, Wong C, Wong C, Nguyen LH, Nguyen MH. 2016. Rates of treatment eligibility in follow-up of patients with chronic hepatitis B (CHB) across various clinical settings who were initially ineligible at presentation. Dig Dis Sci 61:618–625.
View at Publisher | View at Google Scholar - Shankar H, Blanas D, Bichoupan K, Ndiaye D, Carmody E, Martel-Laferriere V, Culpepper-Morgan J, Dieterich DT, Branch AD, Bekele M, Nichols K, Perumalswami PV. 2016. A novel collaborative community-based hepatitis B screening and linkage-to-care program for African immigrants. Clin Infect Dis 62(Suppl 4):S289–S297.
View at Publisher | View at Google Scholar - Xu JJ, Tien C, Chang M, Rhee J, Tien A, Bae HS, Ho FC, Chan LS, Fong TL. 2013. Demographic and serological characteristics of Asian Americans with hepatitis B infection diagnosed at community screenings. J Viral Hepat 20:575–581.
View at Publisher | View at Google Scholar - hang S, Ristau JT, Trinh HN, Garcia RT, Nguyen HA, Nguyen MH. 2012. Undertreatment of Asian chronic hepatitis B patients on the basis of standard guidelines: a community-based study. Dig Dis Sci 57:1373–1383.
View at Publisher | View at Google Scholar - World Health Organisation. 2019. Guidelines for the Prevention, Care and Treatment of Persons with Chronic Hepatitis B Infection. Available at: http://www.who.int/hepatitis/publications/hepatitis-b-guidelines/en/ [Accessed February 22, 2020]
View at Publisher | View at Google Scholar - Marcellin P., Wong D.K., Sievert W., Buggisch P., Petersen J., Flisiak R., Manns M., Kaita K., Krastev Z., Lee S.S. Ten-year efficacy and safety of tenofovir disoproxil fumarate treatment for chronic hepatitis B virus infection. Liver Int. 2019:1868–1875.
View at Publisher | View at Google Scholar - Cho W.H., Lee H.J., Bang K.B., Kim S.B., Song I.H. Development of tenofovir disoproxil fumarate resistance after complete viral suppression in a patient with treatment-naïve chronic hepatitis B: A case report and review of the literature. World J. Gastroenterol. 2018;24:1919–1924.
View at Publisher | View at Google Scholar - Park E.-S., Lee A.R., Kim D.H., Lee J.-H., Yoo J.-J., Ahn S.H., Sim H., Park S., Kang H.S., Won J. Identification of a quadruple mutation that confers tenofovir resistance in chronic hepatitis B patients. J. Hepatol. 2019;70:1093–1102.
View at Publisher | View at Google Scholar - Mokaya J., McNaughton A.L., Bester P.A., Goedhals D., Barnes E., Marsden B.D., Matthews P.C. Hepatitis B virus resistance to tenofovir: fact or fiction? A synthesis of the evidence to date. A systematic literature review and structural analysis of drug resistance mechanisms. 2020;5(151) doi: 10.12688/wellcomeopenres.15992.1.
View at Publisher | View at Google Scholar - Lemoine M., Eholié S., Lacombe K. Reducing the neglected burden of viral hepatitis in Africa: strategies for a global approach. J. Hepatol. 2015;62:469–476.
View at Publisher | View at Google Scholar - Mokaya J., McNaughton A.L., Hadley M.J., Beloukas A., Geretti A.-M., Goedhals D., Matthews P.C. A systematic review of hepatitis B virus (HBV) drug and vaccine escape mutations in Africa: a call for urgent action. PLoS Negl. Trop. Dis. 2018;12 doi: 10.1371/journal.pntd.0006629.
View at Publisher | View at Google Scholar - World Health Organisation . 2016. Combating Hepatitis B and C to Reach Elimination by 2030. Available at: http://apps.who.int/iris/bitstream/10665/206453/1/WHO_HIV_2016.04_eng.pdf?ua=1
View at Publisher | View at Google Scholar - Cooke G.S., Andrieux-Meyer I., Applegate T.L., Atun R., Burry J.R., Cheinquer H., Dusheiko G., Feld J.J., Gore C. Griswold M.G. Accelerating the elimination of viral hepatitis: a Lancet Gastroenterology & Hepatology Commission. Lancet Gastroenterol. Hepatol. 2019;4:135–184.
View at Publisher | View at Google Scholar - World Health Organisation. (2017). Global Hepatitis Report. Available at: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ [Accessed February 22, 2020]
View at Publisher | View at Google Scholar - Maponga T.G., McNaughton A.L., Van Schalkwyk M., Hugo S., Nwankwo C., Taljaard J., Mokaya J., Smith D.A., van Vuuren C., Goedhals D. Treatment advantage in HBV/HIV coinfection compared to HBV monoinfection in a South African cohort. J. Infect. 2020;81:121–130.
View at Publisher | View at Google Scholar - Spearman C.W.N., Sonderup M.W., Botha J.F., van der Merwe S.W., Song E., Kassianides C., Newton K.A., Hairwadzi H.N., Division of Hepatology, Department of Medicine, University of Cape Town, South Africa South African guideline for the management of chronic hepatitis B: 2013. S. Afr. Med. J. 2013;103:337–349.
View at Publisher | View at Google Scholar - World Health Organisation . 2019. Updated Recommendations on First-line and Second-line Antiretroviral Regimens and Post-exposure Prophylaxis and Recommendations on Early Infant Diagnosis of HIV. Available at: http://www.who.int/hiv/pub/guidelines/ARV2018update/en/ [Accessed March 9, 2020]
View at Publisher | View at Google Scholar - McNaughton A.L., Roberts H.E., Bonsall D., de Cesare M., Mokaya J., Lumley S.F., Golubchik T., Piazza P., Martin J.B., de Lara C. Illumina and Nanopore methods for whole genome sequencing of hepatitis B virus (HBV) Sci.
View at Publisher | View at Google Scholar - Rep. 2019;9:7081. artel N., Gomes S.A., Chemin I., Trépo C., Kay A. Improved rolling circle amplification (RCA) of hepatitis B virus (HBV) relaxed-circular serum DNA (RC-DNA) J. Virol. Methods. 2013;193:653–659
View at Publisher | View at Google Scholar
