

Review Article | DOI: https://doi.org/10.31579/2834-8664/075
Dual Mechanisms of Hyperuricemia-Associated Renal Injury: A Systematic Review of Crystal-Dependent and Crystal-Independent Pathways
- Gudisa Bereda *
- Felix Pius Omullo
1 Department of Pharmacy, Rift Valley University, Addis Ababa,1000, Ethiopia.
2 Department of Medical Services, Equity Afya Limited, Lodwar, Turkana County, Kenya.
*Corresponding Author: Gudisa Bereda, Department of Pharmacy, Rift Valley University, Addis Ababa,1000, Ethiopia.
Citation: Gudisa Bereda, Felix Pius Omullo.(2025 Dual Mechanisms of Hyperuricemia-Associated Renal Injury: A Systematic Review of Crystal-Dependent and Crystal-Independent Pathways .International Journal of clinical and Medical Case Reports.4(4); DOI:10.31579/2834-8664/075.
Copyright: © 2025, Gudisa Bereda, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 27 June 2025 | Accepted: 14 July 2025 | Published: 06 August 2025
Keywords: hyperuricemia; renal injury; endothelial dysfunction; crystal-dependent; crystal- independent; systematic review
Abstract
Background: Hyperuricemia, marked by elevated uric acid levels, is associated with renal disorders like acute kidney injury and chronic kidney disease, through both crystal-dependent and crystal- independent mechanisms. Objective: This review aims to evaluate the crystal-dependent and crystal- independent mechanisms by which hyperuricemia induces renal injury. Design: A systematic review of the literature. Participants: Human and animal studies. Measurements: A total of 1549 articles were initially identified from PubMed, Web of Science, Scopus, and Google Scholar. After removing 659 duplicates and screening titles and abstracts, 572 articles were excluded, and 16 could not be retrieved, leaving 302 for full-text review. Of these, 17 studies met the eligibility criteria and were included. Risk of bias was assessed using SYRCLE for animal studies, ROB2 for human studies, and NOS for observational studies. Results: From seventeen studies: nine animal experiments, one human experiment, and seven observational studies. Animal studies showed hyperuricemia causes preglomerular arteriolopathy, glomerular hypertension, and worsens nephrotoxicity. Human studies demonstrated elevated uric acid, even without crystals, activates intrarenal RAS, increases oxidative stress, and reduces nitric oxide. Clinical studies confirmed high uric acid is linked to CKD progression, with very low levels also risky (“J-shaped” relationship). Endothelial dysfunction is a unifying mechanism, promoting inflammation and fibrosis in crystal-dependent injury and vasoconstriction and renal damage in crystal-independent injury. Conclusions: This review confirmed that hyperuricemia damages the kidney through both crystal-dependent and crystal- independent pathways, with endothelial dysfunction as a key mediator. Further human studies are needed to confirm these findings and explore new treatments.
Introduction:
Hyperuricemia, characterized by elevated levels of uric acid in the blood, has long been recognized for its association with various renal disorders, particularly acute kidney injury (AKI) and chronic kidney disease (CKD) [1,2]. The pathogenic role of uric acid in kidney dysfunction is multifaceted, involving complex cellular and molecular pathways that include oxidative stress, inflammation, endothelial dysfunction, and vascular remodeling [3]. Crystal-dependent mechanisms are primarily driven by the deposition of monosodium urate crystals (MSU) crystals in renal tissues, which can lead to direct mechanical damage and trigger inflammatory responses [4]. These crystals can activate the immune system, promote the release of pro-inflammatory cytokines, and stimulate reactive oxygen species (ROS) production, leading to tubular and interstitial injury. On the other hand, crystal-independent mechanisms of hyperuricemia-induced renal injury involve the effects of elevated uric acid levels on renal hemodynamics, endothelial function, and the renin-angiotensin- aldosterone system (RAAS) [5]. Uric acid has been shown to impair the autoregulatory capacity of preglomerular vessels, promote vasoconstriction, and increase glomerular pressure, thereby contributing to glomerular hypertension and further kidney damage [6]. Despite these advances, previous reviews have often examined crystal-dependent and crystal-independent mechanisms separately, lacking an integrated synthesis that connects experimental findings to clinical outcomes. The novel insight of this systematic review lies in its comprehensive approach: it simultaneously evaluates both crystal-dependent and crystal-independent pathways, integrates evidence from animal experimental, human experimental, and observational studies, and emphasizes endothelial dysfunction as a unifying mediator of renal injury. By doing so, this review provides a translational framework linking molecular mechanisms to clinical implications, thereby helping to guide future research and therapeutic strategies for hyperuricemia-associated kidney disease.
Methods:
2.1.Search Strategy:
A comprehensive literature search was conducted across four electronic databases—PubMed, Scopus, Web of Science, and Google Scholar—to identify studies examining the mechanisms of hyperuricemia-induced renal injury. Boolean operators and Medical Subject Headings (MeSH) terms were used to refine the search. The following terms were applied, combining concepts with ʺANDʺ and ʺORʺ: (ʺhyperuricemiaʺ OR ʺuric acidʺ) AND (ʺrenal injuryʺ OR ʺacute kidney injuryʺ OR ʺkidney dysfunctionʺ) AND (ʺmonosodium urate crystalsʺ OR ʺurate crystalsʺ OR ʺcrystal-dependent mechanismsʺ) AND (ʺcrystal-independent mechanismsʺ OR ʺoxidative stressʺ OR ʺvascular dysfunctionʺ OR ʺinflammationʺ) AND (ʺrenal pathophysiologyʺ OR ʺglomerular injuryʺ OR ʺtubulointerstitial fibrosisʺ). Articles published from 2000 to February 2025 were included.
2.2.Inclusion And Exclusion Criteria:
This systematic review followed the PICO framework: animal and human models with hyperuricemia-related renal injury (Patient); hyperuricemia induction methods (Intervention); control groups without induction or with different conditions (Comparison); and dual mechanisms of renal injury, including crystal-dependent and crystal-independent pathways (Outcome). Eligible studies included animal experimental, human experimental, and observational studies that investigated hyperuricemia-induced renal injury, focusing on both crystal-dependent and crystal- independent mechanisms. Only studies published in English were considered. Exclusion criteria included reviews, meta-analyses, commentaries, editorials, letters, studies that did not directly examine the relevant mechanisms, and those with incomplete data or lacking full-text availability.
2.3.Study Selection Process:
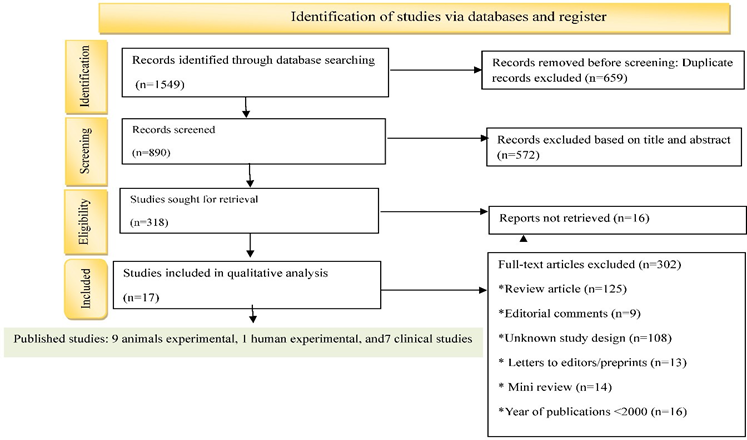
Studies were selected based on their relevance to hyperuricemia-associated renal injury and inclusion of crystal-dependent or crystal-independent mechanisms. A total of 549 articles were initially identified; 189 duplicates were removed, and 272 were excluded based on titles and abstracts, with six articles not retrieved. Additionally, 82 full-text articles were excluded due to being mini- reviews, letters, studies with mixed or unknown designs, outdated publications, or review articles. After detailed evaluation, 17 studies were included in the final qualitative analysis: nine animal experimental studies, one human experimental study, and seven observational studies. The review adhered to PRISMA guidelines [Figure 1][8], and the PRISMA checklist is provided in Table S1 of the Supplementary Appendix.
Figure 1: PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) flow diagram.
2.4.Data Extraction:
Data extraction was performed independently by both authors to ensure accuracy and minimize bias. Using a standardized template, they systematically collected relevant information from each study, including study characteristics (authors, year, design, duration, sample size, and population or animal model), type of hyperuricemia induction, outcomes related to renal injury, and investigated mechanisms (crystal-dependent vs. crystal-independent). Primary outcome measures included kidney histopathology, serum creatinine, markers of inflammation and oxidative stress, and molecular biomarkers. For animal studies, species, strain, dose, and experimental procedures were recorded; for human studies, demographics, comorbidities, uric acid levels, biomarkers, and kidney function measures were extracted. Any discrepancies between the two authors were resolved through discussion and mutual agreement.
2.5.Risk Bias Assessment:
Animal studies were assessed using Systematic Review Centre for Laboratory Animal Experimentation (SYRCLE), which evaluates selection, performance, detection, attrition, reporting, and other biases. Human experimental studies were evaluated with Cochrane Risk of Bias 2 (RoB 2), covering randomization, deviations from interventions, missing data, outcome measurement, and selective reporting. Observational studies were assessed with the Newcastle-Ottawa Scale (NOS), focusing on selection, comparability, and outcome.
2.6.Data Synthesis and Analysis:
Data were synthesized qualitatively, with findings categorized into the two primary mechanisms of hyperuricemia-induced renal injury: crystal-dependent and crystal-independent. Due to heterogeneity in study design, population, and outcomes, a qualitative synthesis was performed, organizing results by study type (animal, human experimental, and observational) and mechanism, highlighting consistencies, discrepancies, and translational relevance. The synthesis also summarized molecular and cellular pathways, emphasizing how these pathways overlap and interact.
Results:
3.1.Study Selection Criteria:
A total of 1549 articles were initially identified from electronic databases, including PubMed, Scopus, Web of Science, and Google Scholar. After removing 659 duplicates and screening for relevance, 572 articles were excluded based on title and abstract, and 16 could not be retrieved, leaving 302 for full-text assessment. Of these, 17 studies met the eligibility criteria and were included in the final analysis [11–20,31–37]. Data from animal experimental, human experimental, and observational studies were analyzed separately to identify consistent patterns, mechanistic insights, and discrepancies. The results were then integrated to examine how crystal-dependent and crystal- independent mechanisms of hyperuricemia contribute to renal injury.
3.2.Study Characteristics:
The 17 studies included in the final analysis comprised nine animal experiments, one human experimental study, and seven observational studies [Table 1]. Animal studies primarily used Sprague-Dawley rats (200–400 g) and Swiss/uricase transgenic mice (30–35 g, and 4 months old), with sample sizes ranging from 6 to 7 animals per group, totaling approximately 70 animals. Study durations ranged from 5 days to 7 weeks. These studies investigated hyperuricemia-induced renal injury, including glomerular hypertension, tubulointerstitial inflammation, endothelial dysfunction, and both crystal-dependent and crystal-independent mechanisms, sometimes using genetic manipulations or uric acid–inducing agents such as oxonic acid [11–19]. The human experimental study included 249 participants and examined uric acid’s effects on intrarenal renin-angiotensin system activation and vascular responses [20]. Observational studies involved 263 to 173,357 participants, totaling approximately 193,000 individuals, and assessed serum uric acid in relation to kidney function, acute kidney injury, chronic kidney disease progression, and mortality, highlighting dose-response and “J-shaped” associations [31–37].
3.3.Risk Of Bias Assessment Of Included Studies:
Across the nine experimental studies, most SYRCLE risk-of-bias domains were rated as ‘some concerns’ due to poor reporting of randomization, allocation concealment, housing and blinding, while baseline group similarity was generally low risk and blinding of caregivers/outcome assessors was high risk; incomplete data were minimal and selective reporting/other biases remained unclear (Figure 2). The RoB-2 assessment of Perlstein TS et al. (2004) indicates some concerns across several domains. The study did not describe its randomization, allocation concealment, blinding procedures, or handling of missing data, all of which raise the potential risk of bias. Although the primary outcome—renal vascular response to Ang II—was an objective measure, the possibility of selective reporting cannot be ruled out. Using the Newcastle–Ottawa Scale, the included observational studies scored between 5 and 8 stars, indicating moderate to high methodological quality (Table 2). Most cohort studies achieved higher scores due to stronger selection and outcome assessment, while cross- sectional studies scored lower, mainly because of limited control for confounding and lack of follow- up.
Table 1: The overview of the eligible study included in this review.
S/N | Authors (Year) | Study Type | Selection | Comparability | Outcome/Exposure |
| Quality |
1 | Joo HJ (2020) [31] | Cross-sectional | ★★ | ★ | ★★ | Moderate | |
2 | San KooB (2021) [32] | Cross-sectional | ★★ | ★★ | ★★ | Moderate | |
3 | Nagore D (2024) [33] | Prospective cohort | ★★★ | ★★ | ★★★ | High | |
4 | Srivastava A (2019) [34] | Prospective cohort | ★★★ | ★★ | ★★ | High | |
5 | Srivastava A (2018) [35] | Prospective cohort | ★★★ | ★★ | ★★ | High | |
6 | Lee EH (2015) [36] | Large single-center observational | ★★★ | ★★ | ★★ | High | |
7 | Kanbay M (2011) [37] | Observational cohort | ★★★ | ★★ | ★★ | High |
Table 2: Standard NOS criteria risk assessment of observational studies.
Legend (Table 2): Selection: representativeness, definition of cases/exposed, selection of controls/non-exposed, ascertainment of exposure (max 4 stars). Comparability: controlled for main confounders (max 2 stars). Outcome/Exposure: assessment, follow-up, ascertainment method (max 3 stars). Quality: High (7–9), Moderate (4–6), Low (0–3).

Figure 2: Risk of Bias Assessment of animal studies by using SYRCLE tool.
3.4.Animal Experimental:
3.4.1.Uric Acid-Associated Renal Injury Via Crystal-Dependent Mechanisms In Animal Experimental Studies
Uric acid can induce AKI through crystal-dependent mechanisms, primarily via MSU crystal precipitation in renal tubules. This leads to tubular obstruction, epithelial injury, inflammation, and fibrosis [9,10]. Sánchez-Lozada LG et al. (2008) demonstrated that UA crystal deposition increases intratubular pressure, impairing autoregulatory capacity in preglomerular vessels and rendering glomerular capillaries more susceptible to systemic hypertension [11]. Sánchez-Lozada LG et al. (2002, 2005) reported that MSU crystals accelerate renal fibrosis in IgA nephropathy, exacerbate oxidative stress and inflammation in cyclosporin nephropathy, and cause preglomerular arteriolopathy with vascular stiffness and hypertrophic remodeling, further elevating glomerular hydrostatic pressure [12,13].Romi MM et al. (2017) highlighted that UA crystals induce endothelial dysfunction and upregulate endothelin-1 (ET-1), promoting vasoconstriction, vascular smooth muscle proliferation, and sustained renal inflammation via NF-κB pathways [14]. Mazzali M et al. (2002) showed that hyperuricemia worsens cyclosporin nephropathy by accelerating tubular injury, inflammatory infiltration, and oxidative stress, leading to progressive fibrosis [15]. Romero F et al. (2009) found that UA administration exacerbated gentamicin-induced nephrotoxicity in rats, where downregulation of MMP-9 impaired extracellular matrix remodeling and promoted tubulointerstitial fibrosis [16]. Kono H et al. (2010) demonstrated that uric acid released from necrotic cells acts as a damage-associated molecular pattern (DAMP), activating the NLRP3 inflammasome and triggering IL-1β and IL-18 production, which amplifies renal inflammation and tubular injury [17]. Rabb H et al. (2000) observed that CD4/CD8-deficient mice exhibited more severe tubular injury and elevated serum creatinine due to impaired T-cell-mediated clearance of uric acid crystals, prolonging inflammation and potentially contributing to CKD progression [18]. Roncal CA et al. (2007) reported that mild hyperuricemia in cisplatin-induced acute renal failure significantly increased tubular injury and inflammation by promoting monocyte chemokine (MCP-1) release and leukocyte infiltration [19]. Crystal-dependent UA mechanisms lead to tubular obstruction, intrarenal inflammation, endothelial dysfunction, and progressive renal injury, with contributions from oxidative stress, immune cell activation, and vascular remodeling, ultimately
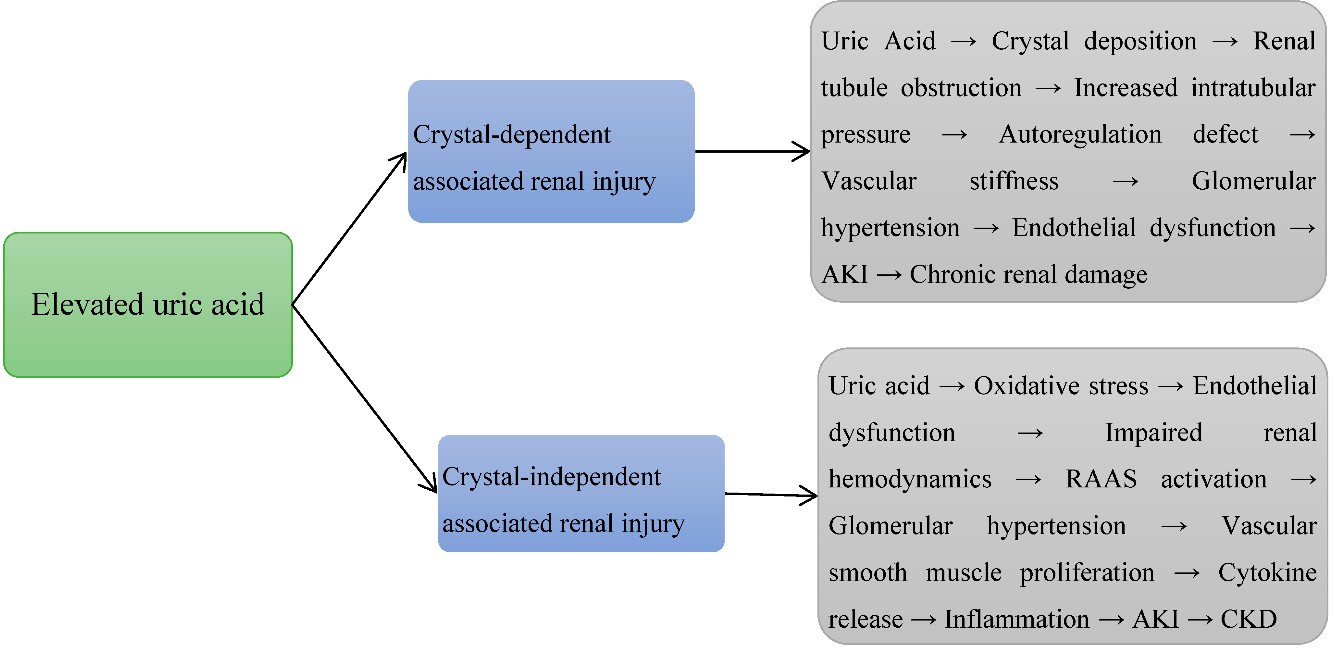 |
linking AKI to CKD progression [3,5] [Figure 3].
Figure 3: Elevated uric acid-induced renal injury via crystal-dependent and crystal independent mechanisms in animal experimental studies.
3.4.2.Findings In Relation To Crystal-Independent Uric Acid-Induced Renal Injury In Animal Experimental Studies
Uric acid (UA) can also induce AKI via crystal-independent mechanisms, including oxidative stress, endothelial dysfunction, inflammation, and impaired renal hemodynamics [2]. Sánchez- Lozada LG (2008) demonstrated that UA disrupts renal autoregulation in preglomerular vessels by increasing oxidative stress, reducing nitric oxide (NO) bioavailability, and triggering endothelial dysfunction, ultimately impairing glomerular filtration [11]. Sánchez-Lozada LG (2002) highlighted UA’s role in activating the renin-angiotensin-aldosterone system (RAAS), exacerbating hypertension, and promoting kidney inflammation and fibrosis in IgA nephropathy and cyclosporin nephropathy [12]. Sánchez-Lozada LG (2005) reported that hyperuricemia induces preglomerular arteriolopathy and glomerular hypertension through vascular smooth muscle proliferation, eNOS suppression, and increased systemic vascular resistance, transmitting hypertension to the glomerulus and aggravating endothelial dysfunction [13]. Romi MM (2017) showed that UA elevates ET-1 expression, promotes vascular smooth muscle proliferation, and activates NF-κB-mediated pro-inflammatory cytokines, collectively impairing renal microcirculation and increasing AKI susceptibility [14]. Mazzali M (2002) reported that UA worsens cyclosporin-induced microvascular and tubulointerstitial injury through oxidative stress, diminished antioxidant defenses, and intensified inflammation [15]. In gentamicin-induced nephrotoxicity models, UA
administration exacerbated tubular injury by downregulating MMP-9,impairing tissue repair, and promoting fibrosis [16]. Kono H (2010) demonstrated that UA acts as a DAMP, activating the NLRP3 inflammasome, enhancing cytokine release, and promoting endothelial dysfunction and mitochondrial impairment, leading to renal vasoconstriction, hypoxia, and tubular apoptosis [17]. Romero F (2009) found that mild hyperuricemia amplified cisplatin-induced renal tubular injury via MCP-1-mediated leukocyte recruitment, worsening inflammation and nephrotoxicity [16]. Studies on T-cell involvement indicated that CD4/CD8-deficient mice exhibited reduced tubular injury and lower serum creatinine levels, suggesting T cells sustain UA-induced inflammation via antigen presentation and cytokine release (IL-1β, TNF-α) [18]. Rabb H (2000) and Roncal CA (2007) highlighted UA’s disruption of autophagy and interference with cellular repair pathways, exacerbating kidney injury [18,19] (Figure 3). Legend (Figure 3): Arrows (→) indicate processes aggravated by uric acid crystal deposition or soluble uric acid, leading to worsening kidney injury.
3.5.Human Experimental:
3.5.1.Uric Acid–Induced Renal Injury Via Crystal-Dependent Mechanisms In Human Experimental Studies:
Mild hyperuricemia has been identified as a factor that worsens cisplatin (CP)-induced acute renal failure (ARF) in humans [20,21]. Perlstein TS (2004) explained that elevated UA levels can lead to the formation of MSU crystals, which precipitate primarily in the kidneys, where uric acid is filtered and excreted [20]. Deposition of UA crystals in renal tubules causes obstruction, increases intratubular pressure, and impairs glomerular filtration rate (GFR), thereby reducing renal function [21]. These crystals also activate inflammatory pathways via NLRP3 inflammasome activation, resulting in the release of pro-inflammatory cytokines, such as IL-1β. This inflammatory cascade recruit’s immune cells, including neutrophils and macrophages, further amplifying tissue injury. Additionally, UA crystals stimulate monocyte chemokines, such as MCP-1, enhancing recruitment of immune cells to sites of renal injury and worsening renal dysfunction [22]. Crystal deposition also contributes to oxidative stress by increasing ROS, leading to apoptosis and necrosis of renal tubular cells, which further amplifies tubular injury and inflammation [23]. Infiltrating neutrophils and macrophages sustain cytokine release, protease activity, and ROS production, creating a vicious cycle that exacerbates renal damage [24,25]. Consequently, renal tubular dysfunction manifests as impaired reabsorption of water, electrolytes, and solutes, culminating in AKI with elevated serum creatinine and reduced urine output, thereby worsening renal function in humans [26] (figure 4).
3.5.2.Uric Acid–Induced Renal Injury Via Crystal-Independent Mechanisms In Human Experimental Studies
Perlstein TS (2004) demonstrated that hyperuricemia can impair kidney function even in the absence of urate crystal deposition, such as in models of chronic pancreatitis (CP)-induced acute ARF [20]. Uric acid contributes to renal tubular injury and inflammation through several mechanisms. Mild hyperuricemia stimulates the production of monocyte chemokines, attracting immune cells, particularly monocytes, which differentiate into macrophages and release pro-inflammatory cytokines, including TNF-α, IL-1, and IL-6, thereby exacerbating tubular damage [20,27].Renal tubules, as the primary sites of injury in AKI, undergo oxidative stress, mitochondrial dysfunction, and cellular injury, leading to impaired function and tubular cell shedding [21]. Furthermore, uric acid promotes endothelial dysfunction, reducing renal perfusion through vasoconstriction and aggravating ischemic injury [28]. In its oxidized form (urate), uric acid increases ROS, causing oxidative damage to membranes, proteins, and DNA, which further impairs renal function [29,30]. These findings indicate that hyperuricemia induces
kidney-specific effects—including inflammation, endothelial dysfunction, and oxidative stress— independent of crystal formation [Figure 4]. Legend (Figure 4): Arrows (→) indicate the processes that are aggravated due to uric acid crystal deposition and its effects, leading to worsening kidney injury.

Figure 4: Elevated uric acid-induced renal injury via crystal-dependent and crystal independent mechanisms in human experimental studies.
3.6.Observational Studies:
3.6.1.Uric Acid-Induced Acute Kidney Injury via Crystal-Dependent Mechanisms in Observational Studies:
The role of uric acid in kidney injury is complex, with both high and low levels potentially affecting renal function [21]. Joo HJ (2020) reported that elevated uric acid levels in a South Korean population were associated with decreased kidney function, likely mediated by crystal deposition, inflammation, oxidative stress, renal vasoconstriction, and fibrosis [31]. San Koo B (2021) found that very low uric acid levels (<2>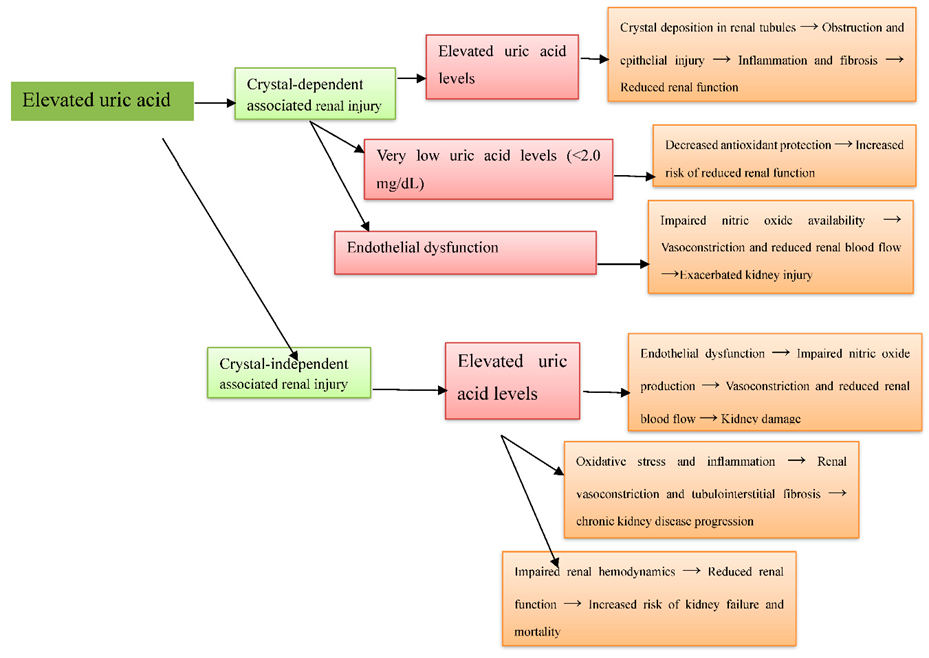
demonstrated that elevated uric acid is independently associated with endothelial dysfunction and reduced eGFR, underlining its contribution to renal injury, which may include crystal-dependent tubular obstruction [37] (Figure 5).
Figure 5: Elevated uric acid-induced renal injury via crystal-dependent and crystal independent mechanisms in observational studies.
3.6.2.Uric Acid-Induced Renal Injury via Crystal-Independent Mechanisms in Observational Studies
Multiple human studies have shown an inverse relationship between elevated uric acid levels and kidney function across diverse populations [38]. Joo HJ (2020) reported that higher uric acid levels in South Koreans were associated with decreased kidney function, likely via inflammation, oxidative stress, renal vasoconstriction, and fibrosis, independent of crystal deposition [31]. San Koo B (2021) found that both high and very low uric acid levels (≤2.0 mg/dL) increased the risk of reduced renal function, potentially through endothelial dysfunction, inflammation, and tubulointerstitial fibrosis, independent of crystal formation [32]. Nagore D (2024) observed that preoperative hyperuricemia did not independently raise the risk of postoperative AKI in cardiac surgery patients, suggesting that hemodynamic and ischemic factors, rather than uric acid itself, were the dominant contributors [33].Srivastava A (2019) showed that uric acid levels did not predict AKI or mortality in critically ill ICU patients, indicating that other contributors such as ischemic injury and multi-organ dysfunction outweigh crystal-independent uric acid effects [34]. Srivastava A (2018) found that higher uric acid levels in CKD patients were linked to increased risk of kidney failure, with a ʺJ-shapedʺ association for all-cause mortality, suggesting systemic effects beyond crystal deposition [35]. Lee EH (2015) reported that moderate preoperative uric acid elevation increased postoperative AKI risk in CABG patients, likely through renal vasoconstriction, inflammation, and endothelial dysfunction, independent of crystals [36]. Kanbay M (2011) demonstrated that elevated uric acid impairs nitric oxide production, causing vasoconstriction and reduced renal perfusion, contributing to kidney injury independently of crystal formation [37] [Figure 5].
Legend (Figure 5): Arrows (→) indicate the processes that are aggravated due to uric acid crystal deposition and its effects, leading to worsening kidney injury.
Discussion:
This systematic review synthesizes evidence on the dual mechanisms of hyperuricemia- associated renal injury—crystal-dependent and crystal-independent pathways—and the central role of endothelial dysfunction in mediating kidney damage. The animal, human experimental, and clinical studies reviewed here collectively demonstrate that uric acid contributes to kidney injury through multiple overlapping mechanisms, extending beyond direct crystal deposition to include vascular and inflammatory pathways.
4.1.Animal Studies
Our systematic review confirms and extends prior work showing that hyperuricemia impairs autoregulation in preglomerular vessels and promotes preglomerular arteriolopathy, resulting in increased glomerular capillary pressure. These findings are consistent with experimental models and human observations, particularly in gout patients [39–41]. Beyond replication, we synthesize evidence that uric acid amplifies nephrotoxicity from agents such as cyclosporine and gentamicin, indicating a synergistic interaction not consistently emphasized in earlier reviews. The vascular changes—preglomerular arteriolopathy and upregulation of endothelin-1 (ET-1)—together with the worsening of drug-induced nephrotoxicity, align with data that uric acid can act as a damage- associated molecular pattern (DAMP), activating the NLRP3 inflammasome and amplifying inflammation and fibrosis [18,19,42]. These combined observations strengthen the biological plausibility of uric acid–driven vascular remodeling and inflammatory injury, while also underscoring the limitations of extrapolating animal data directly to humans because of species differences and model heterogeneity [43,44].
4.2.Human Experimental Studies
Our findings concur with prior evidence that uric acid crystals can directly injure renal tissue by obstructing tubules and activating the NLRP3 inflammasome. Human experimental studies specifically confirm these crystal-dependent effects. Importantly, the review also highlights an independent, crystal-independent role for soluble uric acid: it can activate the intrarenal RAS, increase angiotensin II, raise oxidative stress, and reduce nitric oxide bioavailability—changes that impair endothelial function and promote vasoconstriction, fibrosis, and hypertension [21,45–49]. By emphasizing these vascular effects in the absence of crystals, our synthesis bridges crystal-dependent and crystal-independent pathways and frames uric acid as a systemic (not merely local) nephrotoxin, consistent with findings from both rodent and human models [50–52].
4.3.Observational Studies
Clinical and population studies included in this review demonstrate a generally consistent, dose–response association between higher serum uric acid and reduced kidney function [53–55], a finding supported by Gherghina et al. (2022) and Chang et al. (2010) as evidence that hyperuricemia is an independent risk factor for CKD progression [49,50,55]. Notably, several investigations report a “J-shaped” relationship—Kawasoe et al. (2024) observed that very low uric acid (≤2.0 mg/dL) also correlated with poorer renal outcomes—suggesting that both extremes of uric acid may be harmful [56]. Evidence on specific contexts is mixed: for example, studies of preoperative hyperuricemia and postoperative AKI after CABG conflict (Jiang et al. 2024 found an association, whereas Wang et al. 2018 did not) [46], and other ICU and cohort studies show variable results [57]. Large cohorts and meta-analyses further support hyperuricemia as an independent predictor of adverse renal outcomes [47,51], while other work documents the J-shaped mortality/renal risk pattern [3,56]. These discrepancies likely reflect differences in patient populations, timing of uric acid measurement, and residual confounding. Taken together, the aggregate data link uric acid to renal injury across settings—most plausibly via endothelial dysfunction, altered nitric oxide metabolism, and intrarenal RAS activation—providing mechanistic coherence to the epidemiologic associations [51,58].
4.4.Integrative View
Taken together, this review supports the concept that endothelial dysfunction is the link between crystal-dependent and crystal-independent renal injury. Elevated uric acid impairs nitric oxide bioavailability, increases oxidative stress, and activates the RAS, resulting in vascular remodeling and kidney damage [43,58]. By framing these mechanisms across animal, experimental, and clinical evidence, our review consistently emphasizes the central role of endothelial pathways and introduces asymmetric dimethylarginine (ADMA) as a potential mediator—a feature rarely discussed in previous reviews. These findings highlight that uric acid-induced endothelial dysfunction impairs vasodilation, promotes vascular remodeling, and contributes to renal hypoperfusion and injury, with ADMA acting as an additional inhibitor of endothelial nitric oxide synthase. Collectively, these insights provide a rationale for interventions targeting not only uric acid levels but also oxidative stress, inflammation, and vascular dysfunction.
4.5.Clinical And Research Implications
These findings suggest that interventions should not only lower serum uric acid but also target vascular inflammation, oxidative stress, and RAS activation. Inconsistencies between studies— particularly regarding postoperative AKI and low uric acid levels—emphasize the need for large, well-designed prospective studies with standardized definitions and uniform biomarkers to resolve conflicting evidence and translate experimental findings into clinical practice. Clinically, management of hyperuricemia—through urate-lowering therapy, lifestyle modification, or agents addressing inflammation and RAS activation—may help slow or prevent kidney injury. However, variability in study populations and designs highlights the need for prospective human studies with standardized definitions of hyperuricemia, consistent methods for detecting crystal deposition, and uniform biomarkers of renal injury.
Limitations
This review has several limitations. First, the included studies were heterogeneous in design, encompassing animal experiments, human experiments, and clinical studies, which prevented direct comparisons or meta-analysis. Most of the experimental data were derived from rodent models, and species differences may not fully replicate human renal pathophysiology related to hyperuricemia. Definitions of hyperuricemia, methods of induction, and measures of renal injury varied across studies, and many did not clearly separate crystal-dependent from crystal-independent mechanisms, making it difficult to quantify the contribution of each pathway. In addition, most human evidence was observational rather than longitudinal, limiting causal inference. Publication bias cannot be excluded, as studies with positive findings are more likely to be reported. Finally, because of this heterogeneity and limited data, only a qualitative synthesis was performed, reducing the ability to estimate pooled effects.
Future Directions:
Future research should focus on large, well-designed prospective human studies to clarify the causal relationship between hyperuricemia and renal injury and to clearly differentiate crystal- dependent from crystal-independent pathways. Standardized definitions of hyperuricemia, uniform diagnostic methods for crystal deposition, and consistent biomarkers of renal injury are needed to improve comparability across studies. Mechanistic investigations using advanced imaging, omics approaches, and human tissue models are also required to map the molecular pathways linking uric acid to renal damage. Interventional trials testing urate-lowering therapies or agents targeting inflammation, oxidative stress, or vascular dysfunction could help identify effective strategies to prevent or reverse hyperuricemia-associated kidney injury. Finally, translational studies bridging experimental findings to clinical practice are essential for developing targeted therapies and improving patient outcomes.
Conclusion:
This systematic review highlights the dual mechanisms of hyperuricemia-induced renal injury, integrating both crystal-dependent and crystal-independent pathways, and provides a more comprehensive understanding than previous studies that focused on a single mechanism. Endothelial dysfunction emerges as a central mediator linking both pathways, serving as a unifying mechanism that connects experimental and clinical findings. The review also emphasizes the clinical relevance of a “J-shaped” relationship, suggesting that both high and very low uric acid levels may be detrimental to kidney function. These insights underscore the importance of careful uric acid management in clinical practice and indicate that targeting endothelial dysfunction may offer novel therapeutic strategies to prevent or mitigate renal injury in hyperuricemic patients. Future research should focus on validating these dual mechanisms, targeting endothelial dysfunction, and defining optimal uric acid levels for kidney health.
Acknowledgments:
None.
Informed consent: Written informed consent was obtained from a patient for anonymized patient information to be published in this article.
Declaration Of Conflicting Interests:
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The authors received no financial support for the research, authorship, and/or publication of this article.
Data Availability Statement
Data sharing is not applicable to this article as no data were not created or analyzed in this study.
Credit Authorship Contribution Statement:
Gudisa B: Conceptualization, administration, supervision, methodology, writing – original draft, writing – review and editing. Felix Pius Omullo: Reading, editing, and contribution to methodology. Both authors have read and approved the final version of the manuscript.
References
- Du L, Zong Y, Li H, et al. Hyperuricemia and its related diseases: mechanisms and advances in therapy. Signal Transduction and Targeted Therapy. 2024 Aug 28;9(1):212.
View at Publisher | View at Google Scholar - Zhou M, Huang X, Li R, et al. Association of dietary patterns with blood uric acid concentration and hyperuricemia in northern Chinese adults. Nutrition journal. 2022 Jun 23;21(1):42.
View at Publisher | View at Google Scholar - Ahmad MI, Masood S, Furlanetto DM, et al. Urate crystals; beyond joints. Frontiers in Medicine. 2021 Jun 4; 8:649505.
View at Publisher | View at Google Scholar - Hao G, Xu X, Song J, et al. Lipidomics analysis facilitate insight into the molecular mechanisms of urate nephropathy in a gout model induced by combination of MSU crystals injection and high-fat diet feeding. Frontiers in Molecular Biosciences. 2023 May 3; 10:1190683.
View at Publisher | View at Google Scholar - Tola GB. Exploring the Role of Elevated Uric Acid in Acute Kidney Injury: A Comprehensive Review of Pathways and Therapeutic Approaches.
View at Publisher | View at Google Scholar - Russo E, Verzola D, Cappadona F, et al. The role of uric acid in renal damage-a history of inflammatory pathways and vascular remodeling. Vessel Plus. 2021 Mar 26;5: N-A.
View at Publisher | View at Google Scholar - Sohrabi C, Franchi T, Mathew G, et al. PRISMA 2020 statement: what’s new and the importance of reporting guidelines. Int J Surg 2021; 88:105918.
View at Publisher | View at Google Scholar - Rethlefsen ML, Page MJ. PRISMA 2020 and PRISMA-S: common questions on tracking records and the
View at Publisher | View at Google Scholar - flow diagram. Journal of the Medical Library Association: JMLA. 2022 Apr 1;110(2):253.
View at Publisher | View at Google Scholar - Zhang J, Sun N, Zhang W, et al. The impact of uric acid on musculoskeletal diseases: clinical associations and underlying mechanisms. Frontiers in Endocrinology. 2025 Feb 4; 16:1515176.
View at Publisher | View at Google Scholar - Miake J, Hisatome I, Tomita K, et al. Impact of hyper-and hypo-uricemia on kidney function. Biomedicines. 2023 Apr 24;11(5):1258.
View at Publisher | View at Google Scholar - Sánchez-Lozada LG, Soto V, Tapia E, et al. Role of oxidative stress in the renal abnormalities induced by experimental hyperuricemia. American Journal of Physiology-Renal Physiology. 2008 Oct;295(4): F1134-41.
View at Publisher | View at Google Scholar - Sánchez-Lozada LG, Tapia E, Avila-Casado C, et al. Mild hyperuricemia induces glomerular hypertension in normal rats. American Journal of Physiology-Renal Physiology. 2002 Nov 1;283(5): F1105-10.
View at Publisher | View at Google Scholar - Sanchez-Lozada LG, Tapia E, Santamaria J, et al. Mild hyperuricemia induces vasoconstriction and maintains glomerular hypertension in normal and remnant kidney rats. Kidney international. 2005 Jan 1;67(1):237-47.
View at Publisher | View at Google Scholar - Romi MM, Arfian N, Tranggono U, et al. Uric acid causes kidney injury through inducing fibroblast expansion, Endothelin-1 expression, and inflammation. BMC nephrology. 2017 Dec; 18:1-8.
View at Publisher | View at Google Scholar - Mazzali M, Kanellis J, Han L, et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. American Journal of Physiology-Renal Physiology. 2002 Jun 1;282(6): F991-7.
View at Publisher | View at Google Scholar - Romero F, Pérez M, Chávez M, et al. Effect of uric acid on gentamicin-induced nephrotoxicity in rats–role of matrix metalloproteinases 2 and 9. Basic & clinical pharmacology & toxicology. 2009 Dec;105(6):416-24.7.
View at Publisher | View at Google Scholar - Kono H, Chen CJ, Ontiveros F, et al. Uric acid promotes an acute inflammatory response to sterile cell death in mice. The Journal of clinical investigation. 2010 Jun 1;120(6):1939-49.8.
View at Publisher | View at Google Scholar - Rabb H, Daniels F, O Donnell M, et al. Pathophysiological role of T lymphocytes in renal ischemia- reperfusion injury in mice. American Journal of Physiology-Renal Physiology. 2000 Sep 1;279(3): F525-31.9.
View at Publisher | View at Google Scholar - Roncal CA, Mu W, Croker B, et al. Effect of elevated serum uric acid on cisplatin-induced acute renal failure. American Journal of Physiology-Renal Physiology. 2007 Jan;292(1): F116-22
View at Publisher | View at Google Scholar - Perlstein TS, Gumieniak O, Hopkins PN, et al. Uric acid and the state of the intrarenal renin-angiotensin system in humans. Kidney international. 2004 Oct 1;66(4):1465-70.
View at Publisher | View at Google Scholar - Jung SW, Kim SM, Kim YG, et al. Uric acid and inflammation in kidney disease. American Journal of Physiology-Renal Physiology. 2020 May 11.
View at Publisher | View at Google Scholar - Gu W, Zhao J, Xu Y. Hyperuricemia-induced complications: dysfunctional macrophages serve as a potential bridge. Frontiers in Immunology. 2025 Jan 28; 16:1512093.
View at Publisher | View at Google Scholar - Diaz-Ricart M, Torramade-Moix S, Pascual G, et al. Endothelial damage, inflammation and immunity in chronic kidney disease. Toxins. 2020 Jun 1;12(6):361.
View at Publisher | View at Google Scholar - Yuan Q, Tang B, Zhang C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal transduction and targeted therapy. 2022 Jun 9;7(1):182.
View at Publisher | View at Google Scholar - Gomchok D, Ge RL, Wuren T. Platelets in renal disease. International journal of molecular sciences. 2023 Sep 29;24(19):14724.
View at Publisher | View at Google Scholar - Liu W, Peng J, Wu Y, et al. Immune and inflammatory mechanisms and therapeutic targets of gout: An update. International immunopharmacology. 2023 Aug 1; 121:110466.
View at Publisher | View at Google Scholar - Kimura Y, Tsukui D, Kono H. Uric acid in inflammation and the pathogenesis of atherosclerosis. International journal of molecular sciences. 2021 Jan;22(22):12394.
View at Publisher | View at Google Scholar - Lo CW, Lii CK, Hong JJ, et al. Andrographolide inhibits IL-1β release in bone marrow-derived macrophages and monocyte infiltration in mouse knee joints induced by monosodium urate. Toxicology and applied pharmacology. 2021 Jan 1; 410:115341.
View at Publisher | View at Google Scholar - Kwaifa IK, Bahari H, Yong YK, et al. Endothelial dysfunction in obesity-induced inflammation: molecular mechanisms and clinical implications. Biomolecules. 2020 Feb 13;10(2):291.
View at Publisher | View at Google Scholar - Hénaut L, Candellier A, Boudot C, et al. New insights into the roles of monocytes/macrophages in cardiovascular calcification associated with chronic kidney disease. Toxins. 2019 Sep 12;11(9):529.
View at Publisher | View at Google Scholar - Joo HJ, Kim GR, Choi DW, et al. Uric acid level and kidney function: a cross-sectional study of the Korean national health and nutrition examination survey (2016–2017). Scientific reports. 2020 Dec 10;10(1):21672.
View at Publisher | View at Google Scholar - San Koo B, Jeong HJ, Son CN, et al. J-shaped relationship between chronic kidney disease and serum uric acid levels: a cross-sectional study on the Korean population. Journal of rheumatic diseases. 2021 Oct 1;28(4):225-33.
View at Publisher | View at Google Scholar - Nagore D, Candela A, Bürge M, et al. Uric acid and acute kidney injury in high-risk patients for developing acute kidney injury undergoing cardiac surgery: A prospective multicenter study. Revista Española de Anestesiología y Reanimación (English Edition). 2024 Aug 1;71(7):514-21.
View at Publisher | View at Google Scholar - Srivastava A, Palsson R, Leaf DE, et al. Uric acid and acute kidney injury in the critically ill. Kidney medicine. 2019 Jan 1;1(1):21-30.
View at Publisher | View at Google Scholar - Srivastava A, Kaze AD, McMullan CJ, et al. Uric acid and the risks of kidney failure and death in individuals with CKD. American Journal of Kidney Diseases. 2018 Mar 1;71(3):362-70.
View at Publisher | View at Google Scholar - Lee EH, Choi JH, Joung KW, et al. Relationship between serum uric acid concentration and acute kidney injury after coronary artery bypass surgery. Journal of Korean Medical Science. 2015 Oct 1;30(10):1509-16.
View at Publisher | View at Google Scholar - Kanbay M, Yilmaz MI, Sonmez A, et al. Serum uric acid level and endothelial dysfunction in patients with nondiabetic chronic kidney disease. American journal of nephrology. 2011 Mar 8;33(4):298-304.
View at Publisher | View at Google Scholar - Li X, Meng X, Timofeeva M, et al. Serum uric acid levels and multiple health outcomes: umbrella review of evidence from observational studies, randomised controlled trials, and Mendelian randomisation studies. Bmj. 2017 Jun 7;357.
View at Publisher | View at Google Scholar - Sato Y, Feig DI, Stack AG, et al. The case for uric acid-lowering treatment in patients with hyperuricaemia and CKD. Nature Reviews Nephrology. 2019 Dec;15(12):767-75.
View at Publisher | View at Google Scholar - Sah OS, Qing YX. Associations between hyperuricemia and chronic kidney disease: a review. Nephro- urology monthly. 2015 May 23;7(3): e27233.
View at Publisher | View at Google Scholar - Cui D, Liu S, Tang M, et al. Phloretin ameliorates hyperuricemia-induced chronic renal dysfunction through inhibiting NLRP3 inflammasome and uric acid reabsorption. Phytomedicine. 2020 Jan 1; 66:153111.
View at Publisher | View at Google Scholar - Han YJ, Li S. High levels of uric acid upregulate endothelin receptors: the role of MAPK pathways in an in vitro study. Archives of Medical Science. 2024.
View at Publisher | View at Google Scholar - Ejaz AA, Johnson RJ, Shimada M, et al. The role of uric acid in acute kidney injury. Nephron. 2019 Apr 16;142(4):275-83.
View at Publisher | View at Google Scholar - Wen L, Yang H, Ma L, et al. The roles of NLRP3 inflammasome-mediated signaling pathways in hyperuricemic nephropathy. Molecular and Cellular Biochemistry. 2021 Mar; 476:1377-86.
View at Publisher | View at Google Scholar - Wei X, Zhang M, Huang S, et al. Hyperuricemia: A key contributor to endothelial dysfunction in cardiovascular diseases. The FASEB Journal. 2023 Jul;37(7): e23012.
View at Publisher | View at Google Scholar - Wang M, Lin X, Yang X, et al. Research progress on related mechanisms of uric acid activating NLRP3 inflammasome in chronic kidney disease. Renal failure. 2022 Dec 31;44(1):615-24.
View at Publisher | View at Google Scholar - Galozzi P, Bindoli S, Luisetto R, et al. Regulation of crystal induced inflammation: current understandings and clinical implications. Expert Review of Clinical Immunology. 2021 Jul 3;17(7):773-87
View at Publisher | View at Google Scholar - Zhou Y, Chen M, Zheng J, et al. Insights into the relationship between serum uric acid and pulmonary hypertension. Molecular Medicine Reports. 2023 Nov 21;29(1):10.
View at Publisher | View at Google Scholar - Gherghina ME, Peride I, Tiglis M, et al. Uric acid and oxidative stress—relationship with cardiovascular, metabolic, and renal impairment. International Journal of Molecular Sciences. 2022 Mar 16;23(6):3188.
View at Publisher | View at Google Scholar - Chang HY, Lee PH, Lei CC, et al. Hyperuricemia as an independent risk factor of chronic kidney disease in middle-aged and elderly population. The American journal of the medical sciences. 2010 Jun 1;339(6):509-15.
View at Publisher | View at Google Scholar - Kawasoe S, Kubozono T, Salim AA, et al. J-shaped association between serum uric acid levels and the prevalence of a reduced kidney function: a cross-sectional study using Japanese Health Examination Data. Internal Medicine. 2024 Jun 1;63(11):1539-48.
View at Publisher | View at Google Scholar - Jiang F, Peng Y, Hong Y, et al. Correlation Between Uric Acid/High Density Lipoprotein Cholesterol Ratio and Postoperative AKI in Patients with CABG. International Journal of General Medicine. 2024 Dec 31:6065-74
View at Publisher | View at Google Scholar - Wang JJ, Chi NH, Huang TM, et al. Urinary biomarkers predict advanced acute kidney injury after cardiovascular surgery. Critical care. 2018 Dec; 22:1-3
View at Publisher | View at Google Scholar - Srivastava A, Palsson R, Leaf DE, et al. Uric acid and acute kidney injury in the critically ill. Kidney medicine. 2019 Jan 1;1(1):21-30.
View at Publisher | View at Google Scholar - Weisman A. Associations between allopurinol and cardiovascular and renal outcomes in diabetes. University of Toronto (Canada); 2020.
View at Publisher | View at Google Scholar - Beberashvili I, Sinuani I, Azar A, et al. Serum uric acid as a clinically useful nutritional marker and predictor of outcome in maintenance hemodialysis patients. Nutrition. 2015 Jan 1;31(1):138-47.
View at Publisher | View at Google Scholar - Hahn K, Kanbay M, Lanaspa MA, et al. Serum uric acid and acute kidney injury: a mini review. Journal of advanced research. 2017 Sep 1;8(5):529-36.
View at Publisher | View at Google Scholar - Giordano C, Karasik O, King-Morris K, et al. Uric acid as a marker of kidney disease: review of the current literature. Disease markers. 2015;2015(1):382918.
View at Publisher | View at Google Scholar
